基因组序列比对原理及软件
进行基因的序列比对首先要了解两个概念:
- 同源(homology):两段序列来自于同一个祖先,在后来进化的分化过程中慢慢地各自发生了一些替换或者插入缺失突变,也就是说两段序列不再完全精确的相同。
- 相似(similarity):单纯地指两段序列碱基排序相同,但不具有生物学意义。
同源的序列一定是相似的,但相似的序列不一定是同源的。
如果能事先确定两条序列的同源性再决定是否比对,这样得到的结果会更有意义。
序列比对软件算法分支:
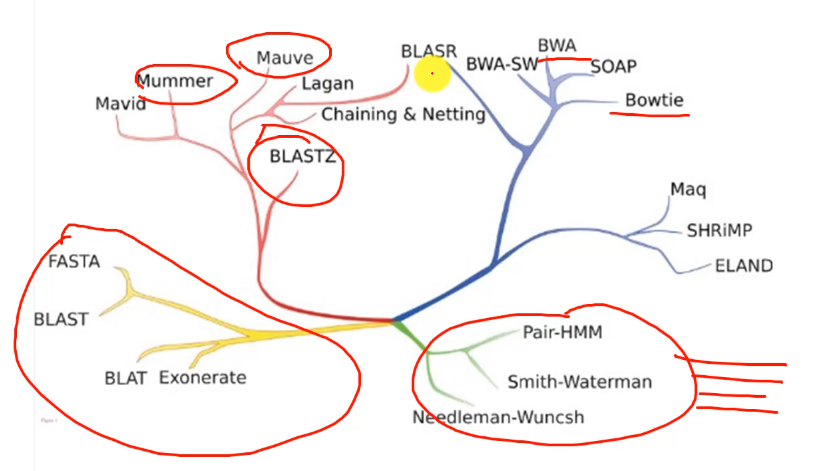
局部比对
两条亲缘关系较远的核酸序列可能只在一些片段上相似,局部比对就可以找到这些局部相似的片段。局部比对允许两条序列不必完整的找到最佳比对方式,只需要使用序列中的某些部分获得最佳匹配得分即可。

局部比对算法更关注于两条序列的相同点,也就是说我们在寻找两条序列的相似性时通常使用局部比对算法(物种分类、基因注释)。
Blast
使用的就是局部比对算法,可以在线比对。常用的功能如下:
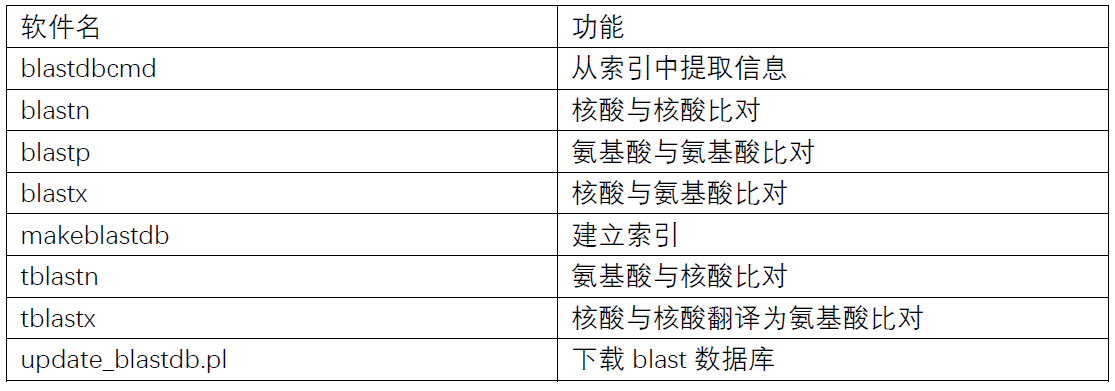
Blast比对最主要的应用就是物种鉴定和基因功能注释。
物种鉴定
拿到一条未知的序列可以直接前往nt库或者nr进行比对来鉴定。
1 | #数据库写前缀就可以 |
基因功能注释
原理也是通过比对已知的序列信息来得到未知的序列中可能包含的信息。所以可以看出比对时所使用的数据库很重要,如果数据库有错误那么比对的结果就也会出错。一般使用nr、COG等数据库。
1 | blastx -db database/nr -query gene.fa -out blastx.out |
Diamond
与blast类似,diamond也是应用于序列比对方面的工具,比对完成之后可以直接导入megan软件进行物种分类以及数据可视化。相比较于blast,diamond的运行速度更快但是只能进行氨基酸水平的比对,不能进行核酸水平的比对。
要注意diamond库与diamond软件的匹配。
1 | #diamond物种鉴定 |
全局比对
衡量两条序列的整体相似性,不关注于局部。全局比对尝试获得两条完整序列之间的最佳比对。
全局比对主要用于比较两个基因组之间的同源性,绘制共线性图等等,也常用于基因组结构变异的检测。

全局比对更加关注于找到两条序列的差异,所以在比较两条序列的差异时我们常使用全局比对。
Mummer(Maximal Unique Matcher)
全局比对软件。该软件集合了一系列功能,例如基因组比对、共线性分析、同源序列查找等。
比对
1 | #比对 |
-q -r参数分别表示仅保留query或者ref上的最佳位置,允许多条ref或者query在另一条上堆叠。-1 则表示取两者的交集(好像得到的结果和-q -r同时输入结果一样)。如下图,-q则保留左2,-r保留右3。
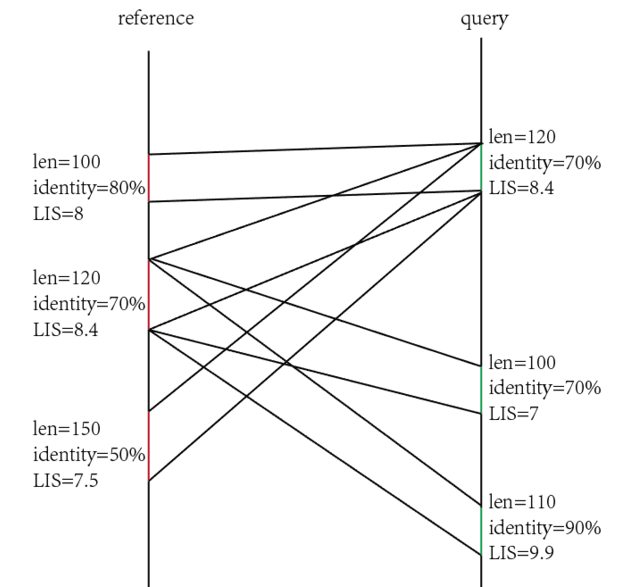
显示比对差异
1 | #显示比对结果 需要给出两条序列在文件中的编号 |
测序数据比对
高通量的测序数据分析一般有两条途径,一条就是将reads拼接得到相对较长的序列,另一条是不经过拼接直接与参考序列比对。由于拼接基因组会消耗较多的计算资源,目前很多分析都是直接使用测序数据比对的方式来分析数据。
测序数据比对的意义:
- 得到每个位点的细节信息:参考序列上的某个位点是否被覆盖到,有多少reads覆盖。
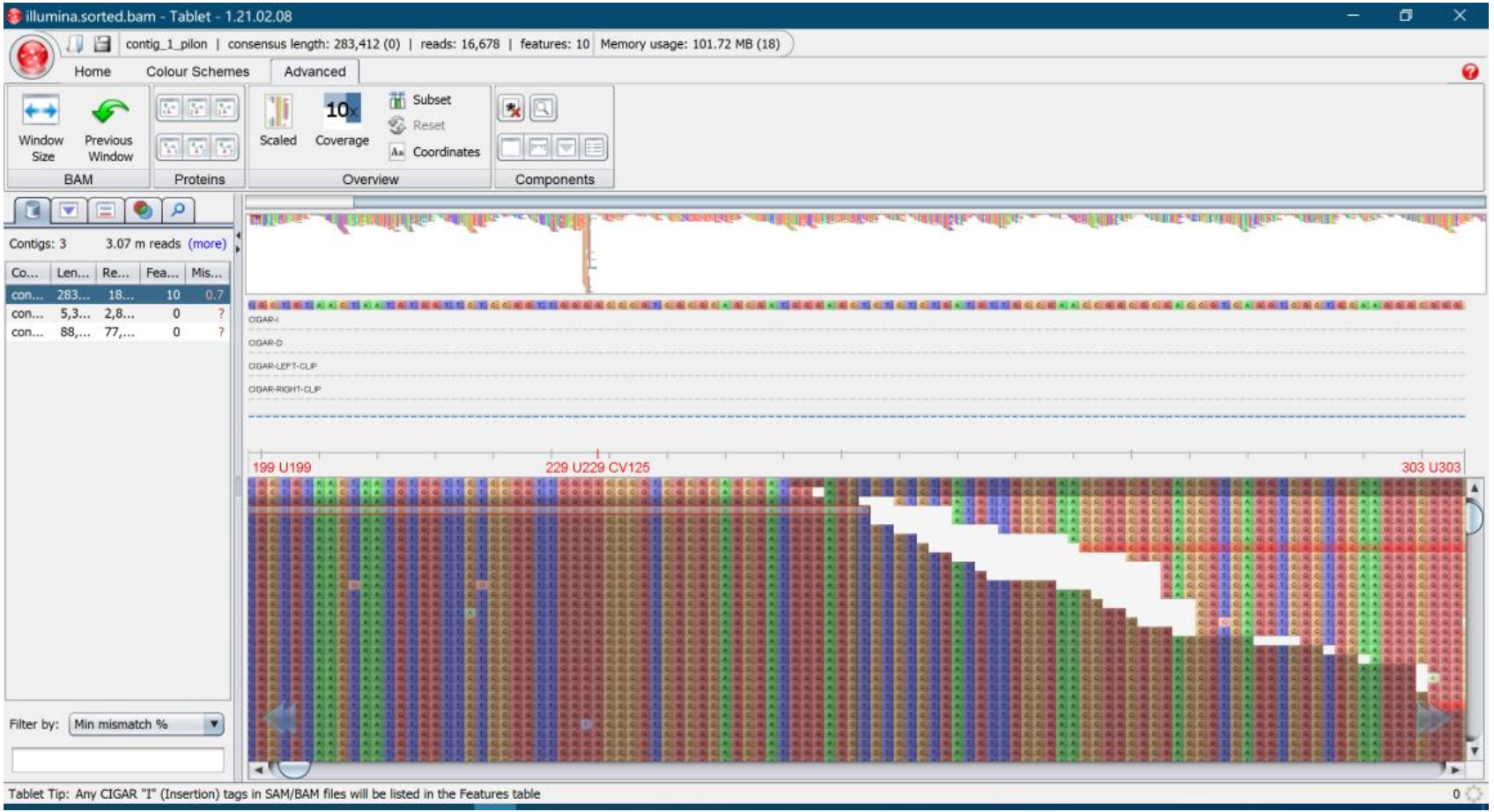
- 变异检测:比较参考序列与测序样品覆盖位点是否一致,如果不一致那么就有可能是潜在的变异位点。
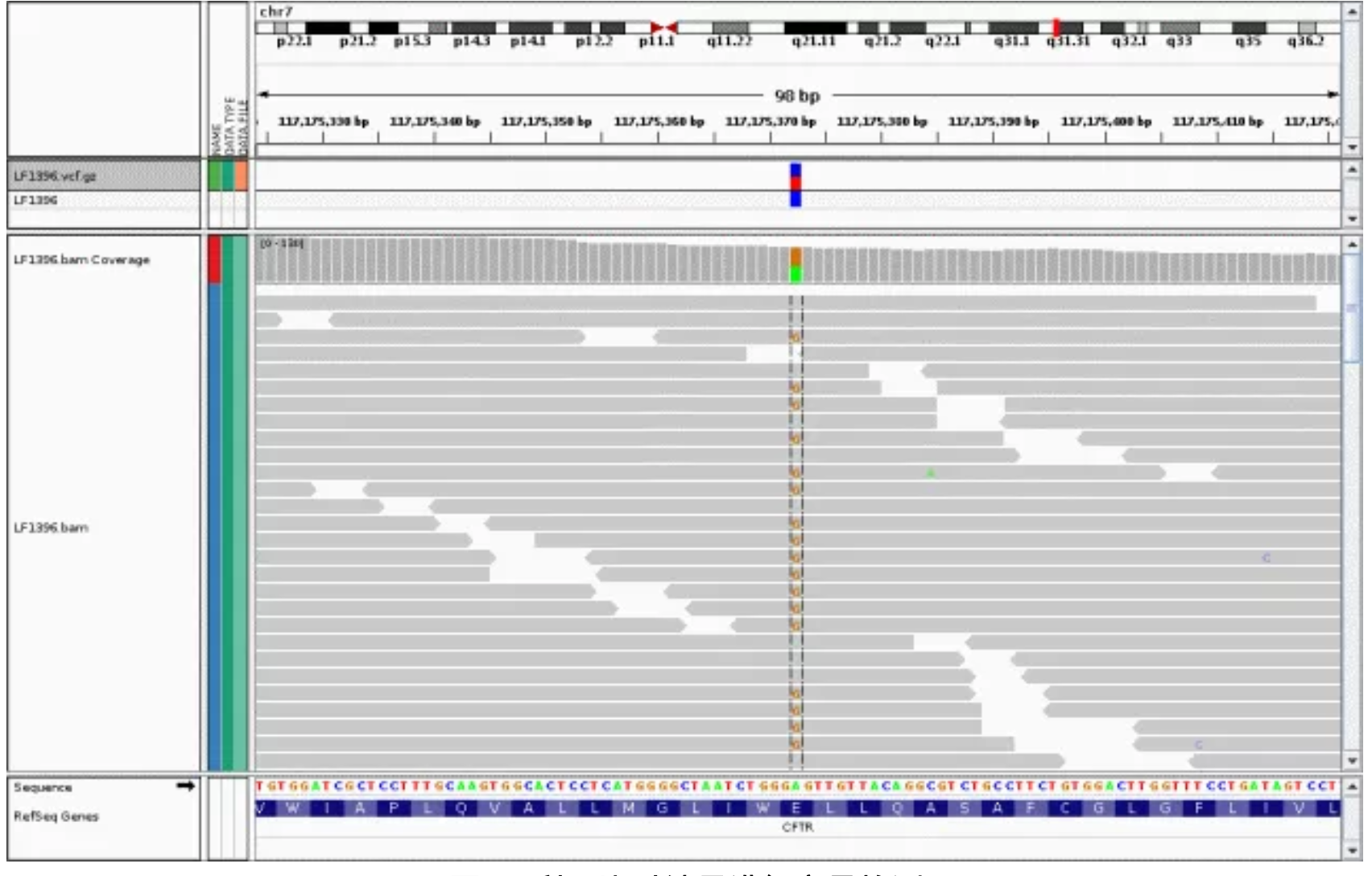
基因表达量计算:将RNAseq测序数据与参考序列进行比较。如两个相同长度的基因A和B,在相同实验测序深度的情况下,A的基因覆盖度100X而B只有50X,则两者基因表达差异为2倍,A基因为高表达。
计算覆盖深度:根据计算每个位点比对上的reads数目可以得到覆盖深度。将全部比对数据除以基因组总长就可以平均覆盖深度。如人基因组中某个染色体的基因覆盖深度出奇的高那么就有可能具有某种染色体疾病(21三体综合症)。
计算覆盖比例:将参考序列被reads覆盖的位点除以位点总数,即可得到覆盖比例。覆盖比例越高证明两者之间的亲缘关系越近。
计算reads利用率:将比对上的reads数除以总reads数。利用率越高说明测序错误越少,样品与参考基因之间的亲缘关系越近。
组装结果纠错:reads与拼接结果比对就是纠错,reads与参考序列比对就是找突变位点。
微生物鉴定:得到测序数据后可以不经过拼接直接与物种分类数据库进行比对用于鉴定微生物。
基因组成环鉴定:如果测序数据可以比对上基因组的首尾两端,则认为基因组成环,成环是细菌基因组是否为完成图的重要标志。
reads之间的overlap关系:三代测序reads与reads之间直接进行比对就可以得到reads之间的overlap关系,用于基因组的拼接。
短序列比对(二代测序)
短序列比对的特点:
- 比对结果是整条比对得上或者比对不上,不能像blast一样分开比对
- 比对仅能允许一定数目的错配和空位
- 序列太短,会出现一条序列比对到多个位置的情况
- 数据量较大时比对比较耗时
比对结果(pair end):
- reads比对不上
- reads比对上
- 单个比对上(或者两者比对上的位置不在正常的insert size 下)
- 双端都能比对上
- 一对一,无错配
- 一对一,有错配
- 一对多,无错配
- 一对多,有错配
bwa(bwa-mem2)比对
1 | #参考序列建立索引 |
长序列比对(三代测序)
随着三代测序技术的发展,目前已经开发出多款适用于三代测序数据的比对软件,如minimap2等。
Minimap2
1 | #minimap2建立索引 |