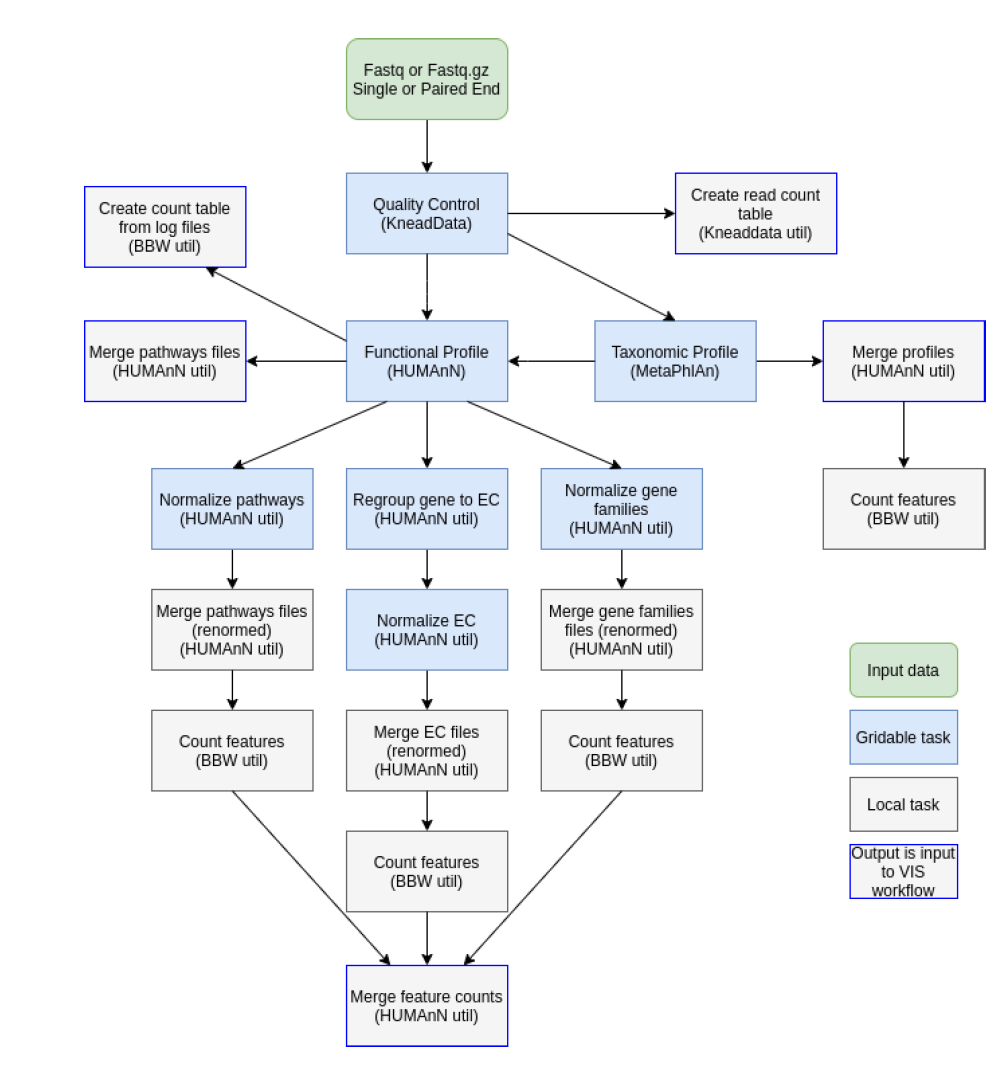
[TOC] # 版本: 1.14, 2022/3/25# 测试环境为Linux Ubuntu 20.04 / CentOS 7.7
一、数据预处理 Data preprocessing
1.1 准备工作 Prepare
- 首次使用请参照
0Install.sh脚本,安装软件和数据库(大约1-3天,仅一次) - 易宏基因组(EasyMetagenome)流程
1Pipeline.sh复制到项目文件夹,如本次为meta - 项目文件夹准备测序数据(seq/*.fq.gz)和样本元数据(result/metadata.txt)
1.1.1 环境变量设置(每次开始分析前必须运行)
设置数据库、软件和工作目录
# Conda软件software安装目录,`conda env list`命令查看,如~/miniconda3soft=/conda2# 公共数据库database(db)位置,如管理员设置/db,个人下载至~/db,并添加其中linux目录中程序至环境变量db=/db# 设置工作目录work directory(wd),如metawd=~/meta# 添加分析所需的软件、脚本至环境变量,添加至~/.bashrc中自动加载PATH=$db/EasyMicrobiome/linux:$db/EasyMicrobiome/script:$PATH# 创建并进入工作目录mkdir -p $wd && cd $wd# 指定某个R语言环境(可选windows下本地运行)alias Rscript="/anaconda2/bin/Rscript --vanilla"
1.1.2 起始文件——序列和元数据
# 创建3个常用子目录:序列,临时文件和结果mkdir -p seq temp result# 上传元数据metadata.txt至result目录,此处下载并重命名wget http://210.75.224.110/github/EasyMetagenome/result/metadata2.txtmv metadata2.txt result/metadata.txt# 检查文件格式,^I为制表符,$为Linux换行,^M$为Windows回车,^M为Mac换行符cat -A result/metadata.txt# 转换Windows回车为Linux换行sed -i 's/\r//' result/metadata.txtcat -A result/metadata.txt
用户使用filezilla上传测序文件至seq目录,本次从其它位置复制,或从网络下载测试数据(多种方法任选其一)
# 方法1. 网络下载测试数据cd seq/awk '{system("wget -c http://210.75.224.110/github/EasyMetagenome/seq/"$1"_1.fq.gz")}' \ <(tail -n+2 ../result/metadata.txt)awk '{system("wget -c http://210.75.224.110/github/EasyMetagenome/seq/"$1"_2.fq.gz")}' \ <(tail -n+2 ../result/metadata.txt)cd ..# 方法2. 从其它目录复制测序数据# cp -rf /db/meta/seq/*.gz seq/# 查看文件大小ls -lsh seq# -l 列出详细信息 (l: list)# -sh 显示人类可读方式文件大小 (s: size; h: human readable)
1.1.3 了解工作目录和文件
显示文件结构
# Ubuntu下安装tree命令# sudo apt install tree# 无法安装请更新软件列表 sudo apt updatetree -L 2# .# ├── pipeline.sh# ├── result# │ └── metadata.txt# ├── seq# │ ├── C1_1.fq.gz# │ ├── C1_2.fq.gz# │ ├── N1_1.fq.gz# │ └── N1_2.fq.gz# └── temp
- 1pipeline.sh是分析流程代码;
- seq目录中有2个样本Illumina双端测序,4个序列文件;
- temp是临时文件夹,存储分析中间文件,结束可全部删除节约空间
- result是重要节点文件和整理化的分析结果图表,
1.2 (可选)FastQC质量评估
# (可选)使用指定位置的(别人安装的)condasource /home/liuyongxin/miniconda2/bin/activate# 启动软件环境conda activate meta# 第一次使用软件要记录软件版本,文章方法中必须写清楚fastqc --version # 0.11.9# time统计运行时间,fastqc质量评估# *.gz为原始数据,-t指定多线程time fastqc seq/*.gz -t 2
质控报告见seq目录,详细解读请阅读《数据的质量控制软件——FastQC》。
multiqc将fastqc的多个报告生成单个整合报告,方法批量查看和比较
# 记录软件版本multiqc --version # 1.8# 整理seq目录下fastqc报告,输出multiqc_report.html至result/qc目录multiqc -d seq/ -o result/qc
查看右侧result/qc目录中multiqc_report.html,单击,选择View in Web Browser查看可交互式报告。
1.3 质量控制
mkdir -p temp/qc
1.3.1 Fastp质量控制环境样品
适用于无宿主污染的环境样品,质控速度快,自动识别接头和低质量,详见:极速的FASTQ文件质控+过滤+校正fastp
# 单样本质控i=C1fastp -i seq/${i}_1.fq.gz -o temp/qc/${i}_1.fastq -I seq/${i}_2.fq.gz -O temp/qc/${i}_2.fastq # 多样本并行tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \ "fastp -i seq/{1}_1.fq.gz -o temp/qc/{1}_1.fastq -I seq/{1}_2.fq.gz -O temp/qc/{1}_2.fastq"
1.3.2 KneadData质控和去宿主
kneaddata是流程,它主要依赖trimmomatic质控和去接头,bowtie2比对宿主,然后筛选非宿主序列用于下游分析 。
详细教程和常见问题,阅读:MPB:随机宏基因组测序数据质量控制和去宿主的分析流程和常见问题
# 记录核心软件版本kneaddata --version # 0.7.4trimmomatic -version # 0.39bowtie2 --version # 2.3.5.1# 可只选一行中部分代码点击Run,如选中下行中#号后面命令查看程序帮助# kneaddata -h # 显示帮助
检查点:zless/zcat查看可压缩文件,检查序列质量格式(质量值大写字母为标准Phred33格式,小写字母为Phred64,需参考附录:质量值转换);检查双端序列ID是否重复,如果重名需要在质控前改名更正。参考附录,质控kneaddata,去宿主后双端不匹配——序列改名。
# 设置某个样本名为变量i,以后再无需修改i=C1# zless查看压缩文件,空格翻页,按q退出。zless seq/${i}_1.fq.gz | head -n4# zcat显示压缩文件,head指定显示行数zcat seq/${i}_2.fq.gz | head -n4
- “|” 为管道符,上一个命令的输出,传递给下一个命令做输入
- gzip: stdout: Broken pipe:管道断开。这里是人为断开,不是错误
- 运行过程中需要仔细阅读屏幕输出的信息
如果序列双端名称一致,改名参见下方代码
(可选) 序列改名,解决NCBI SRA数据双端ID重名问题,详见《MPB:随机宏基因组测序数据质量控制和去宿主的分析流程和常见问题》。
gunzip seq/*.gzsed -i '1~4 s/$/\\1/g' seq/*_1.fqsed -i '1~4 s/$/\\2/g' seq/*_2.fq# 再次核对样本是否标签有重复head seq/C2_1.fqhead seq/C2_2.fq# 结果压缩节省空间gzip seq/*.fq# pigz是并行版的gzip,没装可使用为gzip# pigz seq/*.fq
(可选)单样品质控
若一条代码分割在多行显示时,最好全部选中运行,多行分割的代码行末有一个 “\” 。多行注释命令运行,可全选,按Ctrl+Shift+C进行注释的取消和添加。
- 输入文件:双端FASTQ测序数据,提供给参数-i,seq/${i}_1.fq.gz和 seq/${i}_2.fq.gz
- 参考数据库:宿主基因组索引 -db ${db}/kneaddata/human_genome/hg37dec_v0.1
- 输出文件:质控后的FASTQ测序数据,在目录temp/qc下面,${i}_1_kneaddata_paired_1.fastq和${i}_1_kneaddata_paired_1.fastq,用于后续分析
- 软件位置:
conda env list查看软件安装位置,请务必根据自己软件和数据库安装位置,修改软件trimmomatic和接头文件位置。
(可选)手动设置trimmomatic程序和接头位置
程序目录:${soft}/envs/meta/share/trimmomatic/# 查看multiqc结果中接头污染最严重的C2_1样本,再到fastqc报告中查看接头序列,复制前20个碱基检索确定接头文件grep 'AGATCGGAAGAGCGTCGTGTAGGGAAA' ${soft}/envs/meta/share/trimmomatic/adapters/*# 根据实际情况选择单端SE或双端PE,与原序列比较确定为 TruSeq2-PE.fa,目前多为TruSeq3-PE-2.fa,更准确的是问测序公司要接头文件
100万条序列8线程质控和去宿主,耗时~2m。
i=C1# kneaddata位于liuyongxin中的meta环境,基因组名称为Homo_sapiens或hg37dec_v0.1# soft=/home/liuyongxin/miniconda2/kneaddata -i seq/${i}_1.fq.gz -i seq/${i}_2.fq.gz \ -o temp/qc -v -t 8 --remove-intermediate-output \ --trimmomatic ${soft}/envs/meta/share/trimmomatic/ \ --trimmomatic-options "ILLUMINACLIP:${soft}/envs/meta/share/trimmomatic/adapters/TruSeq2-PE.fa:2:40:15 SLIDINGWINDOW:4:20 MINLEN:50" \ --reorder --bowtie2-options "--very-sensitive --dovetail" \ -db ${db}/kneaddata/human_genome/hg37dec_v0.1# 查看质控后的结果文件大小,确保不是0ls -shtr temp/qc/${i}_1_kneaddata_paired_?.fastq
多样品并行质控
方法1. rush并行管理:注释修改trimmomatic为绝对路径,即修改/home/liuyongxin/miniconda2为你设置的soft变量完整路径,自己用户安装的soft为~/miniconda3(注:存在单引号的代码内不支持变量)
tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \ "kneaddata -i seq/{1}_1.fq.gz -i seq/{1}_2.fq.gz \ -o temp/qc -v -t 3 --remove-intermediate-output \ --trimmomatic $soft/envs/meta/share/trimmomatic/ \ --trimmomatic-options 'ILLUMINACLIP:/home/liuyongxin/miniconda2/envs/meta/share/trimmomatic/adapters/TruSeq2-PE.fa:2:40:15 SLIDINGWINDOW:4:20 MINLEN:50' \ --reorder --bowtie2-options '--very-sensitive --dovetail' \ -db ${db}/kneaddata/human_genome/hg37dec_v0.1"
(可选)方法2. parallel并行质控和去宿主
# 记录软件版本parallel --version # 20160222# 打will cite承诺引用并行软件parallelparallel --citation # parallel软件说明和使用实例# 根据样本列表`:::`并行处理,并行j=2个任务,每个任务t=3个线程,2~7m# 运行下面这行,体会下parallel的工作原理# ::: 表示传递参数;第一个::: 后面为第一组参数,对应于{1};# 第二个::: 后面为第二组参数,对应于{2},依次替换parallel -j 3 --xapply "echo {1} {2}" ::: seq/*_1.fq.gz ::: seq/*_2.fq.gz# --xapply保持文件成对,否则将为两两组合,显示如下:parallel -j 2 "echo {1} {2}" ::: seq/*_1.fq.gz ::: seq/*_2.fq.gz# 从文件列表使用parallel -j 3 --xapply "echo seq/{1}_1.fq.gz seq/{1}_2.fq.gz" ::: `tail -n+2 result/metadata.txt|cut -f1`# 单样本运行成功,且参数设置绝对路径。出现错误`Unrecognized option: -d64`参考**附录,质控Kneaddata,Java版本不匹配——重装Java运行环境**。# 每步分析产生多个文件时新建子文件夹# 每个线程处理百万序列约10分钟,多线程可加速 j x t 倍# 注意此处引物文件必须填写绝对路径,否则无法使用time parallel -j 2 --xapply \ "kneaddata -i seq/{1}_1.fq.gz \ -i seq/{1}_2.fq.gz \ -o temp/qc -v -t 3 --remove-intermediate-output \ --trimmomatic /home/liuyongxin/miniconda2/envs/meta/share/trimmomatic/ \ --trimmomatic-options 'ILLUMINACLIP:/home/liuyongxin/miniconda2/envs/meta/share/trimmomatic/adapters/TruSeq2-PE.fa:2:40:15 SLIDINGWINDOW:4:20 MINLEN:50' \ --reorder --bowtie2-options '--very-sensitive --dovetail' \ -db ${db}/kneaddata/human_genome/Homo_sapiens" \ ::: `tail -n+2 result/metadata.txt|cut -f1`
质控结果改名、临时文件删除和统计
大文件清理,高宿主含量样本可节约>90%空间
rm -rf temp/qc/*contam* temp/qc/*unmatched* temp/qc/*.fqls -l temp/qc/
结果文件链接为新名:awk的system命令批处理系统命令,s为软链(快捷方式)、f为强制(force)
awk '{system("ln -sf `pwd`/temp/qc/"$1"_1_kneaddata_paired_1.fastq temp/qc/"$1"_1.fastq")}' <(tail -n+2 result/metadata.txt)awk '{system("ln -sf `pwd`/temp/qc/"$1"_1_kneaddata_paired_2.fastq temp/qc/"$1"_2.fastq")}' <(tail -n+2 result/metadata.txt)ls -l temp/qc/
质控结果汇总
# 采用kneaddata附属工具kneaddata_read_count_tablekneaddata_read_count_table --input temp/qc \ --output temp/kneaddata.txt# 筛选重点结果列cut -f 1,2,4,12,13 temp/kneaddata.txt | sed 's/_1_kneaddata//' > result/qc/sum.txtcat result/qc/sum.txt# 用R代码统计下质控结果,可在本地运行Rscript -e "data=read.table('result/qc/sum.txt', header=T, row.names=1, sep='\t'); summary(data)"# R转换宽表格为长表格Rscript -e "library(reshape2); data=read.table('result/qc/sum.txt', header=T,row.names=1, sep='\t'); write.table(melt(data), file='result/qc/sum_long.txt',sep='\t', quote=F, col.names=T, row.names=F)"cat result/qc/sum_long.txt# 可用 http://www.ehbio.com/ImageGP/ 绘图展示
1.4 (可选)质控后质量评估
整理bowtie2, trimmomatic, fastqc报告,接头和PCR污染率一般小于1%。结果见:result/qc/multiqc_report_1.html
# 1-2mfastqc temp/qc/*_1_kneaddata_paired_*.fastq -t 2multiqc -d temp/qc/ -o result/qc/# v1.7以后开始使用Python3,v1.8+缺少bowtie2比对结果的统计
二、基于读长分析 Read-based (HUMAnN2)
2.1 准备HUMAnN2输入文件
小技巧:循环批量处理样本列表
# 基于样本元数据提取样本列表命令解析# 去掉表头tail -n+2 result/metadata.txt# 提取第一列样本名tail -n+2 result/metadata.txt|cut -f1# 循环处理样本for i in `tail -n+2 result/metadata.txt|cut -f1`;do echo "Processing "$i; done# ` 反引号为键盘左上角Esc键下面的按键,一般在数字1的左边,代表运行命令返回结果
HUMAnN2要求双端序列合并的文件作为输入,for循环根据实验设计样本名批量双端序列合并。
注意星号和问号,分别代表多个和单个字符。当然大家更不能溜号~~~行分割的代码行末有一个 \
mkdir -p temp/concat# 双端合并为单个文件for i in `tail -n+2 result/metadata.txt|cut -f1`;do cat temp/qc/${i}_?.fastq \ > temp/concat/${i}.fq; done# 查看样品数量和大小ls -shl temp/concat/*.fq# 数据太大,计算时间长,可用head对单端分析截取20M序列,即3G,则为80M行,详见附录:HUMAnN2减少输出文件加速
2.2 HUMAnN2计算物种和功能组成
- 物种组成调用MetaPhlAn2, bowtie2比对至核酸序列,解决有哪些微生物存在的问题;
- 功能组成为humann2调用diamond比对至蛋白库11Gb,解决这些微生物参与哪些功能通路的问题;
- 输入文件:temp/concat/*.fq 每个样品质控后双端合并后的fastq序列
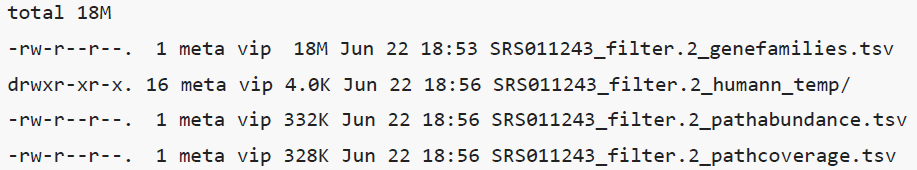
- 输出文件:temp/humann2/ 目录下
- C1_pathabundance.tsv
- C1_pathcoverage.tsv
- C1_genefamilies.tsv
- 整合后的输出:
- result/metaphlan2/taxonomy.tsv 物种丰度表
- result/metaphlan2/taxonomy.spf 物种丰度表(用于stamp分析)
- result/humann2/pathabundance_relab_unstratified.tsv 通路丰度表
- result/humann2/pathabundance_relab_stratified.tsv 通路物种组成丰度表
- stratified(每个菌对此功能通路组成的贡献)和unstratified(功能组成)
启动humann2环境:仅humann2布置于自定义环境下使用
# 方法1. conda加载环境conda activate humann2# 方法2. source加载指定# source /home/liuyongxin/miniconda2/envs/humann2/bin/activate
检查数据库配置是否正确
humann2 --version # v2.8.1humann2_configmkdir -p temp/humann2
单样本1.25M PE150运行测试,8p,2.5M,1~2h;0.2M, 34m;0.1M,30m;0.01M,25m;16p,18m
# CRITICAL ERROR: Can not call software version for bowtie2,见"Perl环境"i=C1# memusg -t humann2 --input temp/concat/${i}.fq --output temp/humann2 --threads 16
多样本并行计算,测试数据约30m,系统耗时12小时
tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \ 'humann2 --input temp/concat/{1}.fq \ --output temp/humann2/'# (可选)大文件清理,humann2临时文件可达原始数据30~40倍# 链接重要文件至humann2目录for i in `tail -n+2 result/metadata.txt|cut -f1`;do ln temp/humann2/${i}_humann2_temp/${i}_metaphlan_bugs_list.tsv temp/humann2/done # 删除临时文件rm -rf temp/concat/* temp/humann2/*_humann2_temp
2.3 物种组成表
2.3.1 样品结果合并
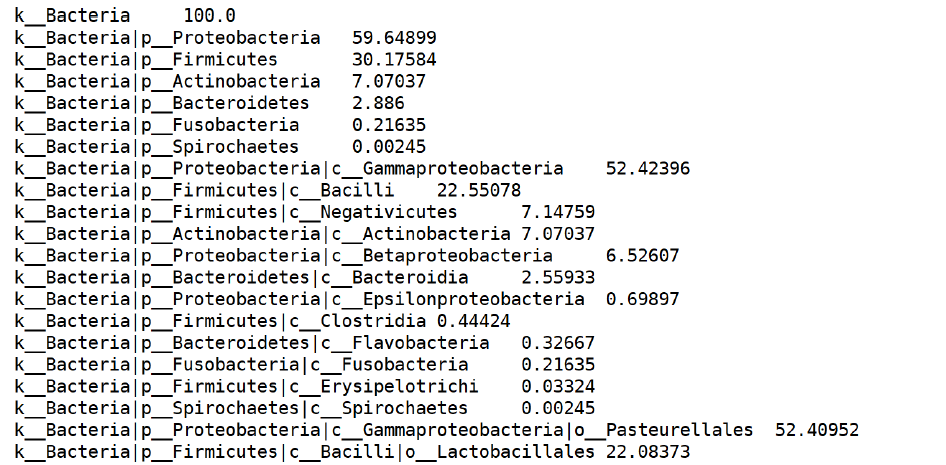
mkdir -p result/metaphlan2# 合并、修正样本名、预览merge_metaphlan_tables.py temp/humann2/*_metaphlan_bugs_list.tsv | \ sed 's/_metaphlan_bugs_list//g' > result/metaphlan2/taxonomy.tsvhead -n5 result/metaphlan2/taxonomy.tsv
2.3.2 转换为stamp的spf格式
metaphlan_to_stamp.pl result/metaphlan2/taxonomy.tsv \ > result/metaphlan2/taxonomy.spfhead -n5 result/metaphlan2/taxonomy.spf# 下载metadata.txt和taxonomy.spf使用stamp分析# 网络分析见附录 metaphlan2-共有或特有物种网络图
2.3.3 (可选)Python绘制热图
# c设置颜色方案,top设置物种数量,minv最小相对丰度,s标准化方法,log为取10为底对数,xy为势图宽和高,图片可选pdf/png/svg格式metaphlan_hclust_heatmap.py \ --in result/metaphlan2/taxonomy.tsv \ --out result/metaphlan2/heatmap.pdf \ -c jet --top 30 --minv 0.1 \ -s log -x 0.4 -y 0.2# 报错解决详见附录:### metaphlan_hclust_heatmap.py报错AttributeError: Unknown property axisbg# 帮助见 metaphlan_hclust_heatmap.py -h# 更多绘制见3StatPlot.sh
2.4 功能组成分析
2.4.1 功能组成合并、标准化和分层
合并通路丰度(pathabundance),含功能和对应物种组成。
可选基因家族(genefamilies 太多),通路覆盖度(pathcoverage)。
注意看屏幕输出# Gene table created: result/humann2/pathabundance.tsv
mkdir -p result/humann2humann2_join_tables \ --input temp/humann2 \ --file_name pathabundance \ --output result/humann2/pathabundance.tsv# 样本名调整:删除列名多余信息head result/humann2/pathabundance.tsvsed -i 's/_Abundance//g' result/humann2/pathabundance.tsv# 预览和统计head result/humann2/pathabundance.tsvcsvtk -t stat result/humann2/pathabundance.tsv
标准化为相对丰度relab(1)或百万比cpm(1,000,000)
humann2_renorm_table \ --input result/humann2/pathabundance.tsv \ --units relab \ --output result/humann2/pathabundance_relab.tsvhead -n5 result/humann2/pathabundance_relab.tsv
分层结果:功能和对应物种表(stratified)和功能组成表(unstratified)
humann2_split_stratified_table \ --input result/humann2/pathabundance_relab.tsv \ --output result/humann2/ # 可以使用stamp进行统计分析
2.4.2 差异比较和柱状图
两样本无法组间比较,在pcl层面替换为HMP数据进行统计和可视化。
参考 https://bitbucket.org/biobakery/humann2/wiki/Home#markdown-header-standard-workflow
- 输入数据:通路丰度表格 result/humann2/pathabundance.tsv
- 输入数据:实验设计信息 result/metadata.txt
- 中间数据:包含分组信息的通路丰度表格文件 result/humann2/pathabundance.pcl
- 输出结果:result/humann2/associate.txt
在通路丰度中添加分组
## 提取样品列表head -n1 result/humann2/pathabundance.tsv | sed 's/# Pathway/SampleID/' | tr '\t' '\n' > temp/header## 对应分组,本示例分组为第2列($2),根据实际情况修改awk 'BEGIN{FS=OFS="\t"}NR==FNR{a[$1]=$2}NR>FNR{print a[$1]}' result/metadata.txt temp/header | tr '\n' '\t'|sed 's/\t$/\n/' > temp/group# 合成样本、分组+数据cat <(head -n1 result/humann2/pathabundance.tsv) temp/group <(tail -n+2 result/humann2/pathabundance.tsv) \ > result/humann2/pathabundance.pclhead -n5 result/humann2/pathabundance.pcl
组间比较,样本量少无差异,结果为4列的文件:通路名字,通路在各个分组的丰度,差异P-value,校正后的Q-value。
演示数据2样本无法统计,此处替换为HMP的结果演示统计和绘图(上传hmp_pathabund.pcl,替换pathabundance.pcl为hmp_pathabund.pcl)。
wget http://210.75.224.110/db/train/meta/result/humann2/hmp_pathabund.pcl# cp /db/humann2/hmp_pathabund.pcl ./mv hmp_pathabund.pcl result/humann2/# 设置输入文件名pcl=result/humann2/hmp_pathabund.pcl# 统计表格行、列数量csvtk -t stat ${pcl}head -n2 ${pcl} |cut -f 1-5# 按分组KW检验,注意第二列的分组列名humann2_associate --input ${pcl} \ --focal-metadatum Group --focal-type categorical \ --last-metadatum Group --fdr 0.05 \ --output result/humann2/associate.txtwc -l result/humann2/associate.txthead -n5 result/humann2/associate.txt
barplot展示通路的物种组成,如:腺苷核苷酸合成
# --sort sum metadata 按丰度和分组排序# 指定差异通路,如 P163-PWY / PWY-3781 / PWY66-409 / PWY1F-823path=PWY-3781humann2_barplot --sort sum metadata \ --input ${pcl} --focal-feature ${path} \ --focal-metadatum Group --last-metadatum Group \ --output result/humann2/barplot_${path}.pdf
2.4.3 转换为KEGG注释
需要下载utility_mapping数据库并配置成功才可以使用。详见软件和数据库安装1soft_db.sh。
支持GO、PFAM、eggNOG、level4ec、KEGG的D级KO等注释,详见humann2_regroup_table -h。
# 转换基因家族为KO(uniref90_ko),可选eggNOG(uniref90_eggnog)或酶(uniref90_level4ec)for i in `tail -n+2 result/metadata.txt|cut -f1`;do humann2_regroup_table \ -i temp/humann2/${i}_genefamilies.tsv \ -g uniref90_ko \ -o temp/humann2/${i}_ko.tsvdone# 合并,并修正样本名humann2_join_tables \ --input temp/humann2/ \ --file_name ko \ --output result/humann2/ko.tsvsed -i '1s/_Abundance-RPKs//g' result/humann2/ko.tsvtail result/humann2/ko.tsv# 与pathabundance类似,可进行标准化renorm、分层stratified、柱状图barplot等操作
2.5 GraPhlAn图
# metaphlan2 to graphlanexport2graphlan.py --skip_rows 1,2 -i result/metaphlan2/taxonomy.tsv \ --tree temp/merged_abundance.tree.txt \ --annotation temp/merged_abundance.annot.txt \ --most_abundant 1000 --abundance_threshold 20 --least_biomarkers 10 \ --annotations 3,4 --external_annotations 7# 参数说明见PPT,或运行 export2graphlan.py --help# graphlan annotationgraphlan_annotate.py --annot temp/merged_abundance.annot.txt \ temp/merged_abundance.tree.txt temp/merged_abundance.xml# output PDF figure, annoat and legendgraphlan.py temp/merged_abundance.xml result/metaphlan2/graphlan.pdf \ --external_legends
2.6 LEfSe差异分析物种
- 输入文件:物种丰度表result/metaphlan2/taxonomy.tsv
- 输入文件:样品分组信息 result/metadata.txt
- 中间文件:整合后用于LefSe分析的文件 result/metaphlan2/lefse.txt,这个文件可以提供给www.ehbio.com/ImageGP 用于在线LefSE分析
- LefSe结果输出:result/metaphlan2/目录下lefse开头和feature开头的文件
前面演示数据仅有2个样本,无法进行差异比较。下面使用result12目录中由12个样本生成的结果表进行演示
# 设置结果目录,自己的数据使用result,演示用result12result=result12# 下载演示数据# wget http://210.75.224.110/db/EasyMetagenome/result12.zip && unzip result12.zip
准备输入文件,修改样本品为组名(可手动修改)
# 预览输出数据head -n3 $result/metaphlan2/taxonomy.tsv# 提取样本行,替换为每个样本一行,修改ID为SampleIDhead -n1 $result/metaphlan2/taxonomy.tsv|tr '\t' '\n'|sed '1 s/ID/SampleID/' >temp/sampleidhead -n3 temp/sampleid# 提取SampleID对应的分组Group(假设为metadata.txt中第二列$2),替换换行\n为制表符\t,再把行末制表符\t替换回换行awk 'BEGIN{OFS=FS="\t"}NR==FNR{a[$1]=$2}NR>FNR{print a[$1]}' $result/metadata.txt temp/sampleid|tr '\n' '\t'|sed 's/\t$/\n/' >groupidcat groupid# 合并分组和数据(替换表头)cat groupid <(tail -n+2 $result/metaphlan2/taxonomy.tsv) > $result/metaphlan2/lefse.txthead -n3 $result/metaphlan2/lefse.txt
方法1. 推荐在线 http://www.ehbio.com/ImageGP 中LEfSe一键分析
方法2. (可选)LEfSe命令行分析代码供参考
# 格式转换为lefse内部格式lefse-format_input.py \ $result/metaphlan2/lefse.txt \ temp/input.in -c 1 -o 1000000# 运行lefse(样本无重复、分组将报错)run_lefse.py temp/input.in \ temp/input.res# 绘制物种树注释差异lefse-plot_cladogram.py temp/input.res \ result/metaphlan2/lefse_cladogram.pdf --format pdf# 绘制所有差异features柱状图lefse-plot_res.py temp/input.res \ $result/metaphlan2/lefse_res.pdf --format pdf # 绘制单个features柱状图# 查看显著差异features,按丰度排序grep -v '-' temp/input.res | sort -k3,3n # 手动选择指定feature绘图,如Firmicuteslefse-plot_features.py -f one \ --feature_name "k__Bacteria.p__Firmicutes" \ --format pdf \ temp/input.in temp/input.res \ $result/metaphlan2/lefse_Firmicutes.pdf# 批量绘制所有差异features柱状图lefse-plot_features.py -f diff \ --archive none --format pdf \ temp/input.in temp/input.res \ $result/metaphlan2/lefse_
2.7 Kraken2物种注释
Kraken2可以快速完成读长(read)层面的物种注释和定量,还可以进行重叠群(contig)、基因(gene)、宏基因组组装基因组(MAG/bin)层面的序列物种注释。
# 方法1.启动kraken2工作环境conda activate kraken2# 方法2.启动指定位置的环境# source /conda2/envs/kraken2/bin/activate# 记录软件版本kraken2 --version # 2.1.1
2.7.1 Kraken2物种注释
{1}代表样本名字
输入:temp/qc/{1}_1_kneaddata_paired*.fastq 质控后的FASTQ数据
参考数据库:-db ${db}/kraken2/mini/,默认标准数据库>50GB,这里使用8GB迷你数据库。
输出结果:每个样本单独输出,temp/kraken2/{1}_report和temp/kraken2/{1}_output
整合后的输出结果: result/kraken2/taxonomy_count.txt 物种丰度表
mkdir -p temp/kraken2
(可选) 单样本注释,5m
i=C1# 1m,--use-mpa-style可直接输出metphlan格式,但bracken无法处理# 2020/12/02版,65K双端序列,38.58%可注释,61.42%未注释,耗时5s,内存峰值8G# 2021/04/23版,65K双端序列,52.17%可注释,52.17%未注释,耗时5s,内存峰值8G# 内存8G的PC可运行,需要与硬盘交换,需3m,内存峰值4.5Gkraken2 --db ${db}/kraken2/mini/ --paired temp/qc/${i}_?.fastq \ --threads 8 --use-names --report-zero-counts \ --report temp/kraken2/${i}.report \ --output temp/kraken2/${i}.output
多样本并行生成report,1样本8线程,内存大但速度快,内存不多不建议用多线程
tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \ "kraken2 --db ${db}/kraken2/mini --paired temp/qc/{1}_?.fastq \ --threads 8 --use-names --report-zero-counts \ --report temp/kraken2/{1}.report \ --output temp/kraken2/{1}.output"
使用krakentools转换report为mpa格式
for i in `tail -n+2 result/metadata.txt|cut -f1`;do kreport2mpa.py -r temp/kraken2/${i}.report \ --display-header \ -o temp/kraken2/${i}.mpa;done
合并样本为表格
mkdir -p result/kraken2# 输出结果行数相同,但不一定顺序一致,要重新排序tail -n+2 result/metadata.txt|cut -f1|rush -j 1 \ 'tail -n+2 temp/kraken2/{1}.mpa | LC_ALL=C sort | cut -f 2 | sed "1 s/^/{1}\n/" > temp/kraken2/{1}_count '# 提取第一样本品行名为表行名header=`tail -n 1 result/metadata.txt | cut -f 1`echo $headertail -n+2 temp/kraken2/${header}.mpa | LC_ALL=C sort | cut -f 1 | \ sed "1 s/^/Taxonomy\n/" > temp/kraken2/0header_counthead -n3 temp/kraken2/0header_count# paste合并样本为表格ls temp/kraken2/*countpaste temp/kraken2/*count > result/kraken2/tax_count.mpa# 检查表格及统计csvtk -t stat result/kraken2/tax_count.mpa
2.7.2 Bracken估计丰度
参数简介:
- -d为数据库,与kraken2一致
- -i为输入kraken2报告文件
- r是读长,此处为100,通常为150
- l为分类级,本次种级别(S)丰度估计,可选域、门、纲、目、科、属、种:D,P,C,O,F,G,S
- t是阈值,默认为0,越大越可靠,但可用数据越少
- -o 输出重新估计的值
循环重新估计每个样品的丰度
# 设置估算的分类级别D,P,C,O,F,G,S,常用 P和Stax=Pmkdir -p temp/brackenfor i in `tail -n+2 result/metadata.txt|cut -f1`;do # i=C1 bracken -d ${db}/kraken2/mini \ -i temp/kraken2/${i}.report \ -r 100 -l ${tax} -t 0 \ -o temp/bracken/${i};done
结果描述:共7列,分别为物种名、ID、分类级、读长计数、补充读长计数、总数、百分比
name taxonomy_id taxonomy_lvl kraken_assigned_reads added_reads new_est_reads fraction_total_readsCapnocytophaga sputigena 1019 S 4798 996 5794 0.23041Capnocytophaga sp. oral taxon 878 1316596 S 239 21 260 0.01034
bracken结果合并成表
# 输出结果行数相同,但不一定顺序一致,要去年表头重新排序# 仅提取第6列reads count,并添加样本名tail -n+2 result/metadata.txt|cut -f1|rush -j 1 \ 'tail -n+2 temp/bracken/{1} | LC_ALL=C sort | cut -f6 | sed "1 s/^/{1}\n/" > temp/bracken/{1}.count '# 提取第一样本品行名为表行名h=`tail -n1 result/metadata.txt|cut -f1`tail -n+2 temp/bracken/${h}|sort|cut -f1 | \ sed "1 s/^/Taxonomy\n/" > temp/bracken/0header.count# 检查文件数,为n+1ls temp/bracken/*count | wc# paste合并样本为表格,并删除非零行paste temp/bracken/*count > result/kraken2/bracken.${tax}.txt# 统计行列,默认去除表头csvtk -t stat result/kraken2/bracken.${tax}.txt
结果筛选
# 需要指定安装R的位置和脚本位置# alias Rscript="/anaconda2/bin/Rscript --vanilla"sd=/db/EasyMicrobiome/script# microbiome_helper按频率过滤,-r可标准化,-e过滤Rscript $sd/filter_feature_table.R \ -i result/kraken2/bracken.${tax}.txt \ -p 0.01 \ -o result/kraken2/bracken.${tax}.0.01# > 0.01(1%)的样本在出现,数量会明显减少csvtk -t stat result/kraken2/bracken.${tax}.0.01# 门水平去除脊索动物grep 'Chordata' result/kraken2/bracken.P.0.01grep -v 'Chordata' result/kraken2/bracken.P.0.01 > result/kraken2/bracken.P.0.01-H# 按物种名手动去除宿主污染,以人为例(需按种水平计算相关结果)# 种水平去除人类P:Chordata,S:Homo sapiensgrep 'Homo sapiens' result/kraken2/bracken.S.0.01grep -v 'Homo sapiens' result/kraken2/bracken.S.0.01 > result/kraken2/bracken.S.0.01-H
分析后清理每条序列的注释大文件
# rm -rf temp/kraken2/*.output
多样性分析/物种组成,详见3StatPlot.sh,Kraken2结果筛选序列见附录
三、组装分析流程 Assemble-based
3.1 拼接 Assembly
3.1.1 MEGAHIT拼接
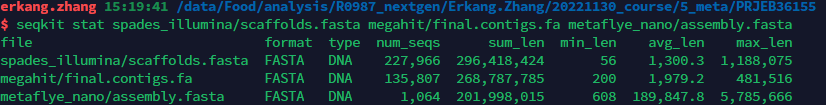
# 启动工作环境conda activate meta# 删除旧文件夹,否则megahit无法运行rm -rf temp/megahit# 组装,10~30m,TB级数据需几天至几周megahit -t 6 \ -1 `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/qc\//;s/$/_1.fastq/'|tr '\n' ','|sed 's/,$//'` \ -2 `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/qc\//;s/$/_2.fastq/'|tr '\n' ','|sed 's/,$//'` \ -o temp/megahit # 统计大小通常300M~5G,如果contigs太多,可以按长度筛选,降低数据量,提高基因完整度,详见附录megahitseqkit stat temp/megahit/final.contigs.fa# 预览重叠群最前6行,前60列字符head -n6 temp/megahit/final.contigs.fa | cut -c1-60# 备份重要结果mkdir -p result/megahit/ln -f temp/megahit/final.contigs.fa result/megahit/# 删除临时文件rm -rf temp/megahit/intermediate_contigs/
# 精细但使用内存和时间更多,15~65mmemusg -t metaspades.py -t 3 -m 100 \ `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/qc\//;s/$/_1.fastq/'|sed 's/^/-1 /'| tr '\n' ' '` \ `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/qc\//;s/$/_2.fastq/'|sed 's/^/-2 /'| tr '\n' ' '` \ -o temp/metaspades# 23M,contigs体积更大seqkit stat temp/metaspades/contigs.fasta# 备份重要结果mkdir -p result/metaspades/ln -f temp/metaspades/contigs.fasta result/metaspades/# 删除临时文件rm -rf temp/metaspades
注:metaSPAdes支持二、三代混合组装,见附录,此外还有OPERA-MS组装二、三代方案
3.1.3 QUAST评估
quast.py result/megahit/final.contigs.fa -o result/megahit/quast -t 2# 生成report文本tsv/txt、网页html、PDF等格式报告# (可选) megahit和metaspades比较quast.py --label "megahit,metapasdes" \ result/megahit/final.contigs.fa \ result/metaspades/contigs.fasta \ -o result/quast# (可选)metaquast评估,更全面,但需下载相关数据库,受网速影响可能时间很长(我很少成功)# metaquast based on silva, and top 50 species genome to accesstime metaquast.py result/megahit/final.contigs.fa -o result/megahit/metaquast
3.2 基因预测、去冗余和定量
# Gene prediction, cluster & quantitfy
# 输入文件:拼装好的序列文件 result/megahit/final.contigs.fa# 输出文件:prodigal预测的基因序列 temp/prodigal/gene.fa# 基因文件大,可参考附录prodigal拆分基因文件,并行计算mkdir -p temp/prodigal# prodigal的meta模式预测基因,35s,>和2>&1记录分析过程至gene.logprodigal -i result/megahit/final.contigs.fa \ -d temp/prodigal/gene.fa \ -o temp/prodigal/gene.gff \ -p meta -f gff > temp/prodigal/gene.log 2>&1 # 查看日志是否运行完成,有无错误tail temp/prodigal/gene.log# 统计基因数量seqkit stat temp/prodigal/gene.fa # 统计完整基因数量,数据量大可只用完整基因部分grep -c 'partial=00' temp/prodigal/gene.fa # 提取完整基因(完整片段获得的基因全为完整,如成环的细菌基因组)grep 'partial=00' temp/prodigal/gene.fa | cut -f1 -d ' '| sed 's/>//' > temp/prodigal/full_length.idseqkit grep -f temp/prodigal/full_length.id temp/prodigal/gene.fa > temp/prodigal/full_length.faseqkit stat temp/prodigal/full_length.fa
3.2.2 基因聚类/去冗余cd-hit
# 输入文件:prodigal预测的基因序列 temp/prodigal/gene.fa# 输出文件:去冗余后的基因和蛋白序列:result/NR/nucleotide.fa, result/NR/protein.famkdir -p result/NR# aS覆盖度,c相似度,G局部比对,g最优解,T多线程,M内存0不限制# 2万基因2m,2千万需要2000h,多线程可加速cd-hit-est -i temp/prodigal/gene.fa \ -o result/NR/nucleotide.fa \ -aS 0.9 -c 0.95 -G 0 -g 0 -T 0 -M 0# 统计非冗余基因数量,单次拼接结果数量下降不大,多批拼接冗余度高grep -c '>' result/NR/nucleotide.fa# 翻译核酸为对应蛋白序列, --trim去除结尾的*seqkit translate --trim result/NR/nucleotide.fa \ > result/NR/protein.fa # 两批数据去冗余使用cd-hit-est-2d加速,见附录
3.2.3 基因定量salmon
# 输入文件:去冗余后的基因和蛋白序列:result/NR/nucleotide.fa# 输出文件:Salmon定量后的结果:result/salmon/gene.count, gene.TPMmkdir -p temp/salmonsalmon -v # 1.4.0# 建索引, -t序列, -i 索引,10ssalmon index \ -t result/NR/nucleotide.fa \ -p 9 \ -i temp/salmon/index # 定量,l文库类型自动选择,p线程,--meta宏基因组模式, 2个任务并行2个样# 注意parallel中待并行的命令必须是双引号,内部变量需要使用原始绝对路径 tail -n+2 result/metadata.txt|cut -f1|rush -j 2 \ "salmon quant \ -i temp/salmon/index -l A -p 3 --meta \ -1 temp/qc/{1}_1.fastq \ -2 temp/qc/{1}_2.fastq \ -o temp/salmon/{1}.quant"# 合并mkdir -p result/salmonsalmon quantmerge --quants temp/salmon/*.quant \ -o result/salmon/gene.TPMsalmon quantmerge --quants temp/salmon/*.quant \ --column NumReads -o result/salmon/gene.countsed -i '1 s/.quant//g' result/salmon/gene.*# 预览结果表格head -n3 result/salmon/gene.*
3.3 功能基因注释
# 输入数据:上一步预测的蛋白序列 result/NR/protein.fa# 中间结果:temp/eggnog/protein.emapper.seed_orthologs# temp/eggnog/output.emapper.annotations# temp/eggnog/output# COG定量表:result/eggnog/cogtab.count# result/eggnog/cogtab.count.spf (用于STAMP)# KO定量表:result/eggnog/kotab.count# result/eggnog/kotab.count.spf (用于STAMP)# CAZy碳水化合物注释和定量:result/dbcan2/cazytab.count# result/dbcan2/cazytab.count.spf (用于STAMP)# 抗生素抗性:result/resfam/resfam.count# result/resfam/resfam.count.spf (用于STAMP)# 这部分可以拓展到其它数据库
3.3.1 基因注释eggNOG(COG/KEGG/CAZy)
# https://github.com/eggnogdb/eggnog-mapper/wiki/eggNOG-mapper-v2# 记录软件版本conda activate eggnogemapper.py --version # 2.1.6# diamond比对基因至eggNOG 5.0数据库, 9p11m, 1~9h,默认diamond 1e-3mkdir -p temp/eggnogtime emapper.py --no_annot --no_file_comments --override \ --data_dir ${db}/eggnog \ -i result/NR/protein.fa \ --cpu 9 -m diamond \ -o temp/eggnog/protein# 比对结果功能注释, 1h # sqlite3.OperationalError: no such table: prots是数据库不配套,重新下载即可emapper.py \ --annotate_hits_table temp/eggnog/protein.emapper.seed_orthologs \ --data_dir ${db}/eggnog \ --cpu 9 --no_file_comments --override \ -o temp/eggnog/output# 2.1较2.0结果又有新变化,添加了#号表头,减少了列sed '1 s/^#//' temp/eggnog/output.emapper.annotations \ > temp/eggnog/outputcsvtk -t headers -v temp/eggnog/output
summarizeAbundance生成COG/KO/CAZy丰度汇总表
mkdir -p result/eggnog# 显示帮助,需要Python3环境,可修改软件第一行指定python位置,如指定某Python执行脚本 /mnt/bai/yongxin/miniconda2/envs/humann3/bin/python3 /db/EasyMicrobiome/script/summarizeAbundance.pysummarizeAbundance.py -h# 汇总,7列COG_category按字母分隔,12列KEGG_ko和19列CAZy按逗号分隔,原始值累加# 指定humann3中的Python 3.7.6运行正常,qiime2中的Python 3.6.13报错summarizeAbundance.py \ -i result/salmon/gene.TPM \ -m temp/eggnog/output \ -c '7,12,19' -s '*+,+,' -n raw \ -o result/eggnog/eggnogsed -i 's/^ko://' result/eggnog/eggnog.KEGG_ko.raw.txtsed -i '/^-/d' result/eggnog/eggnog*# eggnog.CAZy.raw.txt eggnog.COG_category.raw.txt eggnog.KEGG_ko.raw.txt# 添加注释生成STAMP的spf格式,结合metadata.txt进行差异比较awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2} NR>FNR{print a[$1],$0}' \ /db/EasyMicrobiome/kegg/KO_description.txt \ result/eggnog/eggnog.KEGG_ko.raw.txt | \ sed 's/^\t/Unannotated\t/' \ > result/eggnog/eggnog.KEGG_ko.TPM.spf# KO to level 1/2/3summarizeAbundance.py \ -i result/eggnog/eggnog.KEGG_ko.raw.txt \ -m /db/EasyMicrobiome/kegg/KO1-4.txt \ -c 2,3,4 -s ',+,+,' -n raw \ -o result/eggnog/KEGG # CAZyawk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2} NR>FNR{print a[$1],$0}' \ /db/EasyMicrobiome/dbcan2/CAZy_description.txt result/eggnog/eggnog.CAZy.raw.txt | \ sed 's/^\t/Unannotated\t/' > result/eggnog/eggnog.CAZy.TPM.spf# COGawk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2"\t"$3} NR>FNR{print a[$1],$0}' \ /db/EasyMicrobiome/eggnog/COG.anno result/eggnog/eggnog.COG_category.raw.txt > \ result/eggnog/eggnog.COG_category.TPM.spf
3.3.2 (可选)碳水化合物dbCAN2
# 比对CAZy数据库, 用时2~18mmkdir -p temp/dbcan2# --sensitive慢10倍,dbCAN2推荐e值为1e-102,此处结果3条太少,以1e-3为例演示diamond blastp \ --db /db/dbcan2/CAZyDB.09242021 \ --query result/NR/protein.fa \ --threads 9 -e 1e-3 --outfmt 6 --max-target-seqs 1 --quiet \ --out temp/dbcan2/gene_diamond.f6wc -l temp/dbcan2/gene_diamond.f6# 整理比对数据为表格 mkdir -p result/dbcan2# 提取基因与dbcan分类对应表format_dbcan2list.pl \ -i temp/dbcan2/gene_diamond.f6 \ -o temp/dbcan2/gene.list # 按对应表累计丰度,依赖summarizeAbundance.py \ -i result/salmon/gene.TPM \ -m temp/dbcan2/gene.list \ -c 2 -s ',' -n raw \ -o result/dbcan2/TPM# 添加注释生成STAMP的spf格式,结合metadata.txt进行差异比较awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$2} NR>FNR{print a[$1],$0}' \ /db/EasyMicrobiome/dbcan2/CAZy_description.txt result/dbcan2/TPM.CAZy.raw.txt | \ sed 's/^\t/Unannotated\t/' > result/dbcan2/TPM.CAZy.raw.spf# 检查未注释数量,有则需要检查原因# grep 'Unannotated' result/dbcan2/TPM.CAZy.raw.spf|wc -l
3.3.3 抗生素抗性CARD
数据库:https://card.mcmaster.ca/ ,有在线分析平台,本地代码供参考
# 参考文献:http://doi.org/10.1093/nar/gkz935# 软件使用Github: https://github.com/arpcard/rgi# 启动rgi环境conda activate rgirgi -h # 5.2.1# 蛋白注释mkdir -p result/cardcut -f 1 -d ' ' result/NR/protein.fa > temp/protein.fargi main -i temp/protein.fa -t protein \ -n 9 -a DIAMOND --include_loose --clean \ -o result/card/protein
结果说明:
- protein.json,在线可视化
- protein.txt,注释基因列表
3.4 基因物种注释
# Generate report in default taxid outputconda activate metamemusg -t kraken2 --db /db/kraken2/mini \ result/NR/nucleotide.fa \ --threads 3 \ --report temp/NRgene.report \ --output temp/NRgene.output# Genes & taxid listgrep '^C' temp/NRgene.output|cut -f 2,3|sed '1 i Name\ttaxid' \ > temp/NRgene.taxid# Add taxonomyawk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$0} NR>FNR{print $1,a[$2]}' \ /db/EasyMicrobiome/kraken2/taxonomy.txt \ temp/NRgene.taxid \ > result/NR/nucleotide.taxmemusg -t /conda2/envs/humann3/bin/python3 /db/EasyMicrobiome/script/summarizeAbundance.py \ -i result/salmon/gene.TPM \ -m result/NR/nucleotide.tax \ -c '2,3,4,5,6,7,8,9' -s ',+,+,+,+,+,+,+,' -n raw \ -o result/NR/taxwc -l result/NR/tax*|sort -n
四、挖掘单菌基因组/分箱(Binning)
# 主要使用MetaWRAP,演示基于官方测试数据# 主页:https://github.com/bxlab/metaWRAP# 挖掘单菌基因组,需要研究对象复杂度越低、测序深度越大,结果质量越好。要求单样本6GB+,复杂样本如土壤推荐数据量30GB+,至少3个样本# 上面的演示数据12个样仅140MB,无法获得单菌基因组,这里使用官方测序数据演示讲解# 软件和数据库布置需2-3天,演示数据分析过程超10h,标准30G样也需3-30天,由服务器性能决定。
4.1.1 准备数据和环境变量
# 流程: https://github.com/bxlab/metaWRAP/blob/master/Usage_tutorial.md> # 输入数据:质控后的FASTQ序列,文件名格式必须为*_1.fastq和*_2.fastq# C1_1_kneaddata_paired_1.fastq -> C1_1_1.fq# C1_1_kneaddata_paired_2.fastq -> C1_1_2.fq# 放置到 binning/temp/qc 目录下# 拼装获得的contig文件:result/megahit/final.contigs.fa# 放置到 binning/temp/megahit 目录下# # 中间输出文件:# Binning结果:binning/temp/binning# 提纯后的Bin统计结果:binning/temp/bin_refinement/metawrap_50_10_bins.stats# Bin定量结果文件:binning/temp/bin_quant/bin_abundance_heatmap.png# binning/temp/bin_quant/bin_abundance_table.tab (数据表)# Bin物种注释结果:binning/temp/bin_classify/bin_taxonomy.tab# Prokka基因预测结果:binning/temp/bin_annotate/prokka_out/bin.10.ffn 核酸序列# Bin可视化结果:binning/temp/bloblogy/final.contigs.binned.blobplot (数据表)# binning/temp/bloblogy/blobplot_figures (可视化图)# 准备原始数据从头分析,详见公众号或官网教程# 这里我们从质控后数据和拼接结果开始cd ${wd}mkdir -p binning && cd binningmkdir -p temp && cd temp# 这里基于质控clean数据和拼接好的contigs,自己链接自上游分析# 7G质控数据,输入数据文件名格式必须为*_1.fastq和*_2.fastqmkdir -p seqcd seq# 方法1. 下载测序数据# for i in `seq 7 9`;do# wget -c http://210.75.224.110/share/meta/metawrap/ERR01134${i}_1.fastq.gz# wget -c http://210.75.224.110/share/meta/metawrap/ERR01134${i}_2.fastq.gz# done# gunzip *.gz # 解压文件# rename .fq .fastq *.fq # 批量修改扩展名# 方法2. 复制准备好的数据ln -sf ${db}/metawrap/*.fastq ./cd ..# megahit拼接结果mkdir -p megahitcd megahit# wget -c http://210.75.224.110/share/meta/metawrap/final.contigs.fa.gz# gunzip *.gzln -s ${db}/metawrap/*.fa ./cd ../..# 加载运行环境cd ${wd}/binningconda activate metawrap
4.1.2 运行三种分箱软件
metawrap -v# 输入文件为contig和clean reads# 调用三大主流binning程序cococt, maxbin2, metabat2# 8p线程2h,24p耗时1h# nohup 和 & 保证任务在后台不被中断,且记录输出内容到 nohup.out(可选)nohup metawrap binning -o temp/binning -t 1 -a temp/megahit/final.contigs.fa \ --metabat2 --maxbin2 --concoct temp/seq/ERR*.fastq &# 用自己的文件,替换输出文件名为 *1_kneaddata_paired*.fastq # 如果想接上上面的流程使用自己的文件做分析,则把ERR*.fastq替换为 *1_kneaddata_paired*.fastq# 输出文件夹 temp/binning 包括3种软件结果和中间文件
4.1.3 Bin提纯
# 8线程2h, 24p 1hcd ${wd}/binning# rm -rf temp/bin_refinementmetawrap bin_refinement \ -o temp/bin_refinement \ -A temp/binning/metabat2_bins/ \ -B temp/binning/maxbin2_bins/ \ -C temp/binning/concoct_bins/ \ -c 50 -x 10 -t 2# 查看高质量Bin的数量,10个,见temp/bin_refinement/metawrap_50_10_bins.stats目录wc -l temp/bin_refinement/metawrap_50_10_bins.stats# 结果改进程度见temp/bin_refinement/figures/目录
4.1.4 Bin定量
# 使用salmon计算每个bin在样本中相对丰度# 耗时3m,系统用时10m,此处可设置线程,但salmon仍调用全部资源# 需要指定输出文件夹,包括4.3中的参数的输出目录metawrap quant_bins -b temp/bin_refinement/metawrap_50_10_bins -t 8 \ -o temp/bin_quant -a temp/megahit/final.contigs.fa temp/seq/ERR*.fastq# 文件名字改变# 结果包括bin丰度热图`temp/bin_quant/bin_abundance_heatmap.png`# 如果想自己画图,原始数据位于`temp/bin_quant/bin_abundance_table.tab`ls -l temp/bin_quant/bin_abundance_heatmap.png
4.1.5 Bin注释
# Taxator-tk对每条contig物种注释,再估计bin整体的物种,11m (用时66 min)metawrap classify_bins -b temp/bin_refinement/metawrap_50_10_bins \ -o temp/bin_classify -t 2 &# 注释结果见`temp/bin_classify/bin_taxonomy.tab`# export LD_LIBRARY_PATH=/conda2/envs/metagenome_env/lib/:${LD_LIBRARY_PATH} # 这是动态链接库找不到时的一个简单的应急策略ln -s /conda2/envs/metagenome_env/lib/libssl.so.1.0.0 .ln -s /conda2/envs/metagenome_env/lib/libcrypto.so.1.0.0 .# 基于prokka基因注释,4mmetaWRAP annotate_bins -o temp/bin_annotate \ -b temp/bin_refinement/metawrap_50_10_bins -t 1# 每个bin基因注释的gff文件bin_funct_annotations, # 核酸ffn文件bin_untranslated_genes,# 蛋白faa文件bin_translated_genes
多样本受硬件、计算时间限制无法完成时,需要单样本组装、分析。或想进一步提高组装质量,减少污染和杂合度,也可以单样本组装。
参数设定
# 样本名i=ERR011347# 线程数p=1# 任务数j=2# 定义完整度和污染率的阈值(50, 5; Finn NBT 2020; 50, 10, Bowers NBT 2017)c=50x=10
输和文件在seq目录
mkdir -p seqln -s `pwd`/temp/seq/*.fastq seq/
1 megahit组装
单样本并行组装,13m,314m
rm -rf temp/megahit_*time parallel -j ${j} \"metawrap assembly \ -1 seq/{}_1.fastq \ -2 seq/{}_2.fastq \ -o temp/megahit_{} \ -m 100 -t ${p} --megahit" \ ::: `ls seq/|cut -f1 -d '_'|uniq`
2 运行三种bin软件
# 192p, 15m (concoct会使用所有线程)parallel -j ${j} \"metawrap binning \ -o temp/binning_{} -t ${p} \ -a temp/megahit_{}/final_assembly.fasta \ --metabat2 --maxbin2 --concoct \ seq/{}_*.fastq" \::: `ls seq/|cut -f1 -d '_'|uniq`
3 Bin提纯
# 24p,10hparallel -j ${j} \"metawrap bin_refinement \ -o temp/bin_refinement_{} -t ${p} \ -A temp/binning_{}/metabat2_bins/ \ -B temp/binning_{}/maxbin2_bins/ \ -C temp/binning_{}/concoct_bins/ \ -c ${c} -x ${x}" \::: `ls seq/|cut -f1 -d '_'|uniq`
4.2 dRep去冗余种/株基因组集
# 进入虚拟环境,没有用conda安装# conda activate drepsource ${soft}/bin/activate drepcd ${wd}/binning
合并所有bin至同一目录
mkdir -p temp/drep_in# 混合组装分箱并重命名ln -s `pwd`/temp/bin_refinement/metawrap_50_10_bins/bin.* temp/drep_in/rename 'bin' 'mix_all' temp/drep_in/bin.*# 单样品组装分箱结果重命名for i in `ls seq/|cut -f1 -d '_'|uniq`;do ln -s `pwd`/temp/bin_refinement_${i}/metawrap_50_10_bins/bin.* temp/drep_in/ rename "bin." "s_${i}" temp/drep_in/bin.*done# 统计混合和单样本来源数据,10个混,5个单ls temp/drep_in/|cut -f 1 -d '_'|uniq -c# 统计混合批次/单样本来源ls temp/drep_in/|cut -f 2 -d '_'|cut -f 1 -d '.' |uniq -c
按种水平去冗余:15个为10个,8个来自混拼,2个来自单拼
mkdir -p temp/drep95# 15个,40mindRep dereplicate temp/drep95/ \ -g temp/drep_in/*.fa \ -sa 0.95 -nc 0.30 -comp 50 -con 10 -p 3
主要结果:
- 非冗余基因组集:dereplicated_genomes/*.fa
- 聚类信息表:data_tables/Cdb.csv
- 聚类和质量图:firgures/clustering
(可选)按株水平汇总
# 20-30minmkdir -p temp/drep95dRep dereplicate temp/drep95/ \ -g temp/drep_in/*.fa \ -sa 0.99 -nc 0.30 -comp 50 -con 10 -p 24
4.3 GTDB-tk物种注释和进化树
启动软件所在虚拟环境
# gtdbtk与drep安装在了同一个环境# conda activate gtdbtk
细菌基因组物种注释
以上面鉴定的10个种为例,注意扩展名要与输入文件一致,可使用压缩格式gz。主要结果文件描述:此9个细菌基因组,结果位于tax.bac120开头的文件,如物种注释 tax.bac120.summary.tsv。古菌结果位于tax.ar122开头的文件中。
mkdir -p temp/gtdb_classify# 10个基因组,24p,100min 152 G内存gtdbtk classify_wf \ --genome_dir temp/drep95/dereplicated_genomes \ --out_dir temp/gtdb_classify \ --extension fa \ --prefix tax \ --cpus 10
多序列对齐结果建树
# 以9个细菌基因组的120个单拷贝基因建树,1smkdir -p temp/gtdb_infergtdbtk infer \ --msa_file temp/gtdb_classify/tax.bac120.user_msa.fasta \ --out_dir temp/gtdb_infer \ --prefix tax \ --cpus 2
树文件可使用iTOL在线美化,也可使用GraphLan本地美化。
4.4 table2itol制作树注释文件
以gtdb-tk物种注释(tax.bac120.summary.tsv)和drep基因组评估(Widb.csv)信息为注释信息
mkdir -p result/itol# 制作分类学表tail -n+2 temp/gtdb_classify/tax.bac120.summary.tsv|cut -f 1-2|sed 's/;/\t/g'|sed '1 s/^/ID\tDomain\tPhylum\tClass\tOrder\tFamily\tGenus\tSpecies\n/' \ > result/itol/tax.txt# 基因组评估信息sed 's/,/\t/g;s/.fa//' temp/drep95/data_tables/Widb.csv|cut -f 1-7,11|sed '1 s/genome/ID/' \ > result/itol/genome.txt# 整合注释文件awk 'BEGIN{OFS=FS="\t"} NR==FNR{a[$1]=$0} NR>FNR{print $0,a[$1]}' result/itol/genome.txt result/itol/tax.txt|cut -f 1-8,10- > result/itol/annotation.txt
table2itol制作注释文件
cd result/itol/# 设置脚本位置db=/disk1/db/script/table2itol/#db=/db## 方案1. 分类彩带、数值热图、种标签# -a 找不到输入列将终止运行(默认不执行)-c 将整数列转换为factor或具有小数点的数字,-t 偏离提示标签时转换ID列,-w 颜色带,区域宽度等, -D输出目录,-i OTU列名,-l 种标签替换ID# Fatal error: ??????'./table2itol-master/table2itol.R': ?????????Rscript ${db}/table2itol.R -a -c double -D plan1 -i ID -l Species -t %s -w 0.5 annotation.txt# 生成注释文件中每列为单独一个文件## 方案2. 数值柱形图,树门背景色,属标签Rscript ${db}/table2itol.R -a -d -c none -D plan2 -b Phylum -i ID -l Genus -t %s -w 0.5 annotation.txt## 方案3.分类彩带、整数为柱、小数为热图Rscript ${db}/table2itol.R -c keep -D plan3 -i ID -t %s annotation.txt## 方案4. 将整数转化成因子生成注释文件Rscript ${db}/table2itol.R -a -c factor -D plan4 -i ID -l Genus -t %s -w 0 annotation.txt
4.5 PROKKA单菌基因组功能注释
conda activate metawrapexport PERL_5LIB=${PERL5LIB}:${soft}/envs/metawrap/lib/perl5/site_perl/5.22.0/i=bin1time prokka result/contig/${db}.fa \ --kingdom Archaea,Bacteria --cpus 9 \ --outdir temp/prokka/${db}
附录:常见分析问题和经验
质控KneadData
双端序列质控后是否配对的检查
双端序列质控后序列数量不一致是肯定出错了。但即使序列数量一致,也可能序列不对。在运行metawrap分箱时会报错。可以kneaddata运行时添加–reorder来尝试解决。以下提供了检查双端序列ID是否配对的比较代码
# 文件i=sample1seqkit seq -n -i temp/qc/${i}_1_kneaddata_paired_1.fastq|cut -f 1 -d '/' | head > temp/header_${i}_1seqkit seq -n -i temp/qc/${i}_1_kneaddata_paired_2.fastq|cut -f 1 -d '/' | head > temp/header_${i}_2cmp temp/header_${i}_?
Perl环境不匹配
报错’perl binaries are mismatched’的解决
e=~/miniconda3/envs/metaPERL5LIB=${e}/lib/5.26.2:${e}/lib/5.26.2/x86_64-linux-thread-multi
Java不匹配——重装Java运行环境
若出现错误 Unrecognized option: -d64,则安装java解决:
conda install -c cyclus java-jdk
读长分析HUMAnN2
HUMAnN2减少输出文件加速
HUMAnN2是计算非常耗时的步骤,如果上百个10G+的样本,有时需要几周至几月的分析。以下介绍两种快速完成分析,而且结果变化不大的方法。替换下面for循环为原文中的“双端合并为单个文件”部分代码
方法1. 软件分析不考虑双端信息,只用一端可获得相近结果,且速度提高1倍。链接质控结果左端高质量至合并目录
for i in `tail -n+2 result/metadata.txt|cut -f1`;do ln -sf `pwd`/temp/qc/${i}_1_kneaddata_paired_1.fastq temp/concat/${i}.fqdone
方法2. 控制标准样比对时间。测序数据量通常为650G,同一样本分析时间可达10h100h,严重浪费时间而浪费硬盘空间。
可用head对单端分析截取20M序列,即3G,则为80M行
for i in `tail -n+2 result/metadata.txt|cut -f1`;do head -n80000000 temp/qc/${i}_1_kneaddata_paired_1.fastq > temp/concat/${i}.fqdone
正常在首次运行时,会自动下载数据库。有时会失败,解决方法:
方法1. 使用软件安装的用户运行一下程序即可下载成功
方法2. 将我们预下载好的数据索引文件,链接到软件安装目录
db=~/dbsoft=~/miniconda2mkdir -p ${soft}/bin/db_v20ln -s ${db}/metaphlan2/* ${soft}/bin/db_v20/mkdir -p ${soft}/bin/databasesln -s ${db}/metaphlan2/* ${soft}/bin/databases/
CRITICAL ERROR: Can not call software version for bowtie2
解决问题思路:
查看文件位置是否处在conda环境中:type bowtie2。如果不在需要手动设置环境变量的顺序,如果位置正确如在(~/miniconda2/envs/humann2/bin/bowtie2),请往下看;
检测bowtie2运行情况:bowtie2 -h,报错wd.c: loadable library and perl binaries are mismatched (got handshake key 0xde00080, needed 0xed00080)。 错误原因为Perl库版本错误,检查Perl库位置:echo $PERL5LIB,错误原因没有指向环境,并手动修改perl库位置
# 设置你环境变量位置,最好用绝对路径e=~/miniconda2/envs/humann2PERL5LIB=${e}/lib/5.26.2:${e}/lib/5.26.2/x86_64-linux-thread-multi
在网上搜索,axisbg和axis_bgcolor为过时的函数,新版为facecolor,修改为新名称即可 (参考:https://blog.csdn.net/qq_41185868/article/details/81842971)
# 定位文件绝对路径file=`type metaphlan_hclust_heatmap.py|cut -f 2 -d '('|sed 's/)//'`# 替换函数名称为新版sed -i 's/axisbg/facecolor/g' $file
awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {for(i=9;i<=NF;i++) a[i]=$i; print "Tax\tGroup"} \ else {for(i=9;i<=NF;i++) if($i>0.05) print "Tax_"FNR, a[i];}}' \ result/metaphlan2/taxonomy.spf > result/metaphlan2/taxonomy_highabundance.tsv awk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {print "Tax\tGrpcombine";} else a[$1]=a[$1]==""?$2:a[$1]$2;}END{for(i in a) print i,a[i]}' \ result/metaphlan2/taxonomy_highabundance.tsv > result/metaphlan2/taxonomy_group.tsvcut -f 2 result/metaphlan2/taxonomy_group.tsv | tail -n +2 | sort -u >groupfor i in `cat group`; do printf "#%02x%02x%02x\n" $((RANDOM%256)) $((RANDOM%256)) $((RANDOM%256)); done >colorcodepaste group colorcode >group_colorcodeawk 'BEGIN{OFS=FS="\t"}ARGIND==1{a[$1]=$2;}ARGIND==2{if(FNR==1) {print $0, "Grpcombinecolor"} else print $0,a[$2]}' \ group_colorcode result/metaphlan2/taxonomy_group.tsv > result/metaphlan2/taxonomy_group2.tsvawk 'BEGIN{OFS=FS="\t"}{if(FNR==1) {print "Tax",$1,$2,$3,$4, $5, $6, $7, $8 } else print "Tax_"FNR, $1,$2,$3,$4, $5,$6, $7, $8}' \ result/metaphlan2/taxonomy.spf > result/metaphlan2/taxonomy_anno.tsv
生物标志鉴定LEfSe
lefse-plot_cladogram.py:Unknown property axis_bgcolor
若出现错误 Unknown property axis_bgcolor,则修改lefse-plot_cladogram.py里的ax_bgcolor替换成facecolor即可。
# 查看脚本位置,然后使用RStudio或Vim修改type lefse-plot_cladogram.py
物种分类Kraken2
合并样本为表格combine_mpa.py
krakentools中combine_mpa.py,需手动安装脚本,且结果还需调整样本名
combine_mpa.py \ -i `tail -n+2 result/metadata.txt|cut -f1|sed 's/^/temp\/kraken2\//;s/$/.mpa/'|tr '\n' ' '` \ -o temp/kraken2/combined_mpa
提取非植物33090和动物(人)33208序列、选择细菌2和古菌2157
mkdir -p temp/kraken2_qcparallel -j 3 \ "/db/script/extract_kraken_reads.py \ -k temp/kraken2/{1}.output \ -r temp/kraken2/{1}.report \ -1 temp/qc/{1}_1_kneaddata_paired_1.fastq \ -2 temp/qc/{1}_1_kneaddata_paired_2.fastq \ -t 33090 33208 --include-children --exclude \ --max 20000000 --fastq-output \ -o temp/kraken2_qc/{1}_1.fq \ -o2 temp/kraken2_qc/{1}_2.fq" \ ::: `tail -n+2 result/metadata.txt|cut -f1`
组装Megahit
序列长度筛选
megahit默认>200,可选 > 500 / 1000 bp,并统计前后变化;如此处筛选 > 500 bp,序列从15万变为3.5万条,总长度从7M下降到3M
mv temp/megahit/final.contigs.fa temp/megahit/raw.contigs.faseqkit seq -m 500 temp/megahit/raw.contigs.fa > temp/megahit/final.contigs.faseqkit stat temp/megahit/raw.contigs.faseqkit stat temp/megahit/final.contigs.fa
数据太大导致程序中断
报错信息:126 - Too many vertices in the unitig graph (8403694648 >= 4294967294), you may increase the kmer size to remove tons
解决方法:需要增加k-mer,如最小k-mer改为29,不行继续增加或将数据分批次组装
添加参数: –k-min 29 –k-max 141 –k-step 20
二三代混合组装
# 3G数据,耗时3hi=SampleAtime metaspades.py -t 48 -m 500 \ -1 seq/${i}_1.fastq -2 seq/${i}L_2.fastq \ --nanopore seq/${i}.fastq \ -o temp/metaspades_${i}
二三代混合组装OPERA-MS
结果卡在第9步polishing,可添加–no-polishing参数跳过此步;短序列只支持成对文件,多个文件需要cat合并
二三代混合组装
perl ../OPERA-MS.pl \ --short-read1 R1.fastq.gz \ --short-read2 R2.fastq.gz \ --long-read long_read.fastq \ --no-ref-clustering \ --num-processors 32 \ --out-dir RESULTS
二代组装+三代优化
perl ~/soft/OPERA-MS/OPERA-MS.pl \ --contig-file temp/megahit/final.contigs.fa \ --short-read1 R1.fastq.gz \ --short-read2 R2.fastq.gz \ --long-read long_read.fastq \ --num-processors 32 \ --no-ref-clustering \ --no-strain-clustering \ --no-polishing \ --out-dir temp/opera
结果可用quast或seqkit stat统计对二代组装的改进效果
基因序列prodigal
序列拆分并行预测基因
(可选)以上注释大约1小时完成1M个基因的预测。加速可将contigs拆分,并行基因预测后再合并。
# 拆分contigs,按1M条每个文件n=10000seqkit split result/megahit/final.contigs.fa -s $n# 生成拆分文件序列列表ls result/megahit/final.contigs.fa.split/final.contigs.part_*.fa|cut -f 2 -d '_'|cut -f 1 -d '.' \ > temp/split.list# 9线程并行基因预测,此步只用单线程且读写强度不大time parallel -j 9 \ "prodigal -i result/megahit/final.contigs.fa.split/final.contigs.part_{}.fa \ -d temp/gene{}.fa \ -o temp/gene{}.gff -p meta -f gff \ > temp/gene{}.log 2>&1 " \ ::: `cat temp/split.list`# 合并预测基因和gff注释文件cat temp/gene*.fa > temp/prodigal/gene.facat temp/gene*.gff > temp/prodigal/gene.gff
基因去冗余cd-hit
两批基因合并cd-hit-est-2d
cd-hit-est-2d 两批次构建非冗余基因集
A和B基因集,分别有M和N个非冗余基因,两批数据合并后用cd-hit-est去冗余,计算量是(M + N) X (M + N -1)
cd-hit-est-2d比较,只有M X N的计算量
# 计算B中特有的基因cd-hit-est-2d -i A.fa -i2 B.fa -o B.uni.fa \ -aS 0.9 -c 0.95 -G 0 -g 0 \ -T 96 -M 0 -d 0# 合并为非冗余基因集cat A.fa B.uni.fa > NR.fa
cd-hit合并多批基因salmon索引时提示ID重复
# [error] In FixFasta, two references with the same name but different sequences: k141_2390219_1. We require that all input records have a unique name up to the first whitespace (or user-provided separator) character.# 错误解决mv temp/NRgene/gene.fa temp/NRgene/gene.fa.bak# 15G,2m,4Gseqkit rename temp/NRgene/gene.fa.bak -o temp/NRgene/gene.fa
基因定量salmon
找不到库文件liblzma.so.0
- 报错信息:error while loading shared libraries: liblzma.so.0
- 问题描述:直接运行salmon报告,显示找不到lib库,
- 解决方法:可使用程序完整路径解决问题,
alias salmon="${soft}/envs/metagenome_env/share/salmon/bin/salmon"
基因功能数据库
综合功能注释KEGG描述整理
脚本位于 /db/script 目录,https://www.kegg.jp/kegg-bin/show_brite?ko00001.keg 下载htext,即为最新输入文件 ko00001.keg
kegg_ko00001_htext2tsv.pl -i ko00001.keg -o ko00001.tsv
抗生素抗性CARD
# 使用3.1.0和3.1.2均有警告,修改序列名至纯字母数数字也无效# WARNING 2021-07-08 08:58:00,478 : Exception : <class 'KeyError'> -> '5141' -> Model(1692) missing in database. Please generate new database.# WARNING 2021-07-08 08:58:00,478 : Exception : <class 'KeyError'> -> '5141' -> Model(1692)# WARNING 2021-07-08 08:58:00,479 : tetM ---> hsp.bits: 60.8 <class 'float'> ? <class 'str'>
抗生素抗性ResFam
数据库:http://www.dantaslab.org/resfams
参考文献:http://doi.org/10.1038/ismej.2014.106
mkdir -p temp/resfam result/resfam# 比对至抗生素数据库 1mtime diamond blastp \ --db ${db}/resfam/Resfams-proteins \ --query result/NR/protein.fa \ --threads 9 --outfmt 6 --sensitive \ -e 1e-5 --max-target-seqs 1 --quiet \ --out temp/resfam/gene_diamond.f6# 提取基因对应抗性基因列表cut -f 1,2 temp/resfam/gene_diamond.f6 | uniq | \ sed '1 i Name\tResGeneID' > temp/resfam/gene_fam.list# 统计注释基因的比例, 488/19182=2.5%wc -l temp/resfam/gene_fam.list result/salmon/gene.count # 按列表累计丰度summarizeAbundance.py \ -i result/salmon/gene.TPM \ -m temp/resfam/gene_fam.list \ -c 2 -s ',' -n raw \ -o result/resfam/TPM# 结果中添加FAM注释,spf格式用于stamp分析awk 'BEGIN{FS=OFS="\t"} NR==FNR{a[$1]=$4"\t"$3"\t"$2} NR>FNR{print a[$1],$0}' \ ${db}/resfam/Resfams-proteins_class.tsv result/resfam/TPM.ResGeneID.raw.txt \ > result/resfam/TPM.ResGeneID.raw.spf
细菌基因组物种注释GTDB
菌的文件名不要存在非字母数字的符号,否则运行会报错。
# ERROR: ['BMN5'] are not present in the input list of genome to process,但并无此菌,可能是名称 中存在"-"或".",替换为i# 修改metadatased 's/-/i/;s/\./i/' result/metadatab.txt > result/metadata.txt# 修改文件名awk 'BEGIN{OFS=FS="\t"}{system("mv temp/antismash/"$1".fna temp/antismash/"$2".fna")ll }' <(paste result/metadatab.txt result/metadata.txt|tail -n+2)
版本更新记录
1.08 2020.7.20
- KneadData提供数据预处理双端标签唯一命令,兼容最新版;
- 提供HUMAnN3测试版的安装和分析流程(附录1);
- eggNOG升级为emapper 2.0和eggNOG 5.0流程,结果列表从13列变为22列,新增CAZy注释。emapper 1.0版本见附录2。
1.09 2020.10.16
- 新增二、三代混合组装OPERA-MS软件使用 (31Megahit)
- 新增eggNOG-mapper结果COG/KO/CAZy整理脚本summarizeAbundance.py,删除旧版Shell+R代码 (32Annotation)
- 新增MetaWRAP单样本分箱流程 (33Binning)
- 新增dRep实现基因组去冗余 (34Genomes)
- 新增GTDB-Tk基因组物种注释和进化树构建 (34Genomes)
1.10 2021.1.22
- 增加删除中间文件部分,节约空间,防止硬盘写满;
- 正文的补充分析方法、常见问题移至附录,按软件名、问题/方法分级索引;
- 软件使用前,增加检查软件版本命令,方便文章方法中撰写准确版本;
- 删除不稳定的humann3、过时的eggnog版本教程;
- 增加kraken2新环境, 增加bracken, krakentools新工具;
- kraken2结果新增beta多样性PCoA,物种组成堆叠柱状图;
- 增metaspades二、三代组装代码示例;
- 新增KEGG层级注释整理代码;
- 更新dbCAN2中2018版为2020版;
- 新增CARD本地分析流程;
1.11 2021.5.7
- 增加prodigal基因预测并行版方法,使用seqkit split拆分后并行,数10倍加速单线程基因预测步骤;
- 增加megahit拼装结果片段大小选择步骤,使用seqkit -m按长度筛选,并统计筛选前后变化;
- 不常用或可选代码调整到附录
- 两批数据快速合并去冗余cd-hit-est-2d
- 二三代混合组装OPERA-MS的混装和3代优化代码
1.12 2021.8.20
- 新增并行管理软件rush,比parallel更易安装,绿色版无依赖关系,整合在db/linux/目录中
- 新增seqkit,可以统计序列数据量,支持序列长度过滤,格式转换等;
- 新增质控软件fastp,软件fastqc更快,适合单独质控不去宿主;
- kraken2新数据库,同样大小下注释率提高明显;
- eggNOG软件和数据库配套升级
- GTDB-tk软件和数据库需要配套重新才可使用新版25万基因组数据库
1.13 2021.11.19
- 陈同参与EasyMicrobiome的更新,并提交了mac版本代码
- 新增humann2运行bowtie2出错的解决方案
1.14 2022.3.25
- EasyMicrobiome升级为1.14
- 升级miniconda2为miniconda3
- dbcan2从2020/7/31的808M更新为2021/9/24版1016M,格式变化,配套format_dbcan2list.pl更新
- 新增eggnog环境,包含emapper 2.1.6,summarizeAbundance.py含pandas (conda install sklearn-pandas),配套更新数据库
- rgi更新到最新版及配套代码
]]>



.png)