宏基因组测序及分析流程
二代测序数据拼接
质控完成之后,宏基因组二代测序的数据拼接软件使用基本和单菌拼接保持一致,目前大部分软件都支持宏基因组的拼接模式。
SPAdes
该软件支持宏基因组的拼接模式,只需要加上参数–meta即可。
1 | spades.py --meta -1 read_1.fastq.gz -2 read_2.fastq.gz -o spades_result -t 24 |
megahit
该软件运行速度快于SPAdes且占用的计算资源也比较少,但组装效果不如前者。
1 | megahit -o megahit -1 read_1.fastq.gz -2 read_2.fastq.gz -t 24 |
三代测序数据拼接
flye
质控后,三代测序宏基因组数据仍然可以使用flye进行组装,它其中有一个metaflye的模块专门用来组装宏基因组数据。
1 | flye --nano-raw nano.fastq.gz -g 5m -t 12 -o metaflye_result --meta |
值得注意的是在进行完数据拼接后还可以使用纠错软件进行纠错(pilon、racon等),这里可能会涉及到使用二代测序数据辅助纠错。
二代三代软件拼接结果对比
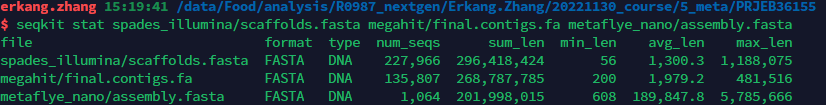
这里可以很明显的看到同为二代测序拼接,megahit所拼接出的contigs不如SPAdes长。Nanopore三代测序在读长方面的优势还是比较明显的。
宏基因组物种分类
物种分类数据库
当前宏基因组物种分类的方法主要就是使用NCBI的物种分类数据库进行比对分类。将测序得到的序列和数据库(如nt库、nr库、refseq库)中的序列进行比对,如果两者具有很好的相似性则认为两者为同源序列,具有共同的祖先。
- nt库为Nucleotide Sequence Database,即核酸序列数据库,其中含有所有目前已知的核酸序列(基因组),是有冗余的。
- nr库为Non-Redundant Protein Sequence Database,即非冗余蛋白库(只包含基因预测的蛋白而不是基因组)。一般DIAMOND比对使用这个数据库。
- refseq库是NCBI官方验证的可以代表某个物种的参考序列所组成的数据库,是非冗余的,一般物种分类就是使用这个。
- taxonomy数据库。这个库里的内容主要是物种的名字和种系,这些物种都至少在遗传数据库中有一条核酸或蛋白序列。其目的是为序列数据库建立一个一致的种系发生分类学,物种分类也需要这个。
物种鉴定软件
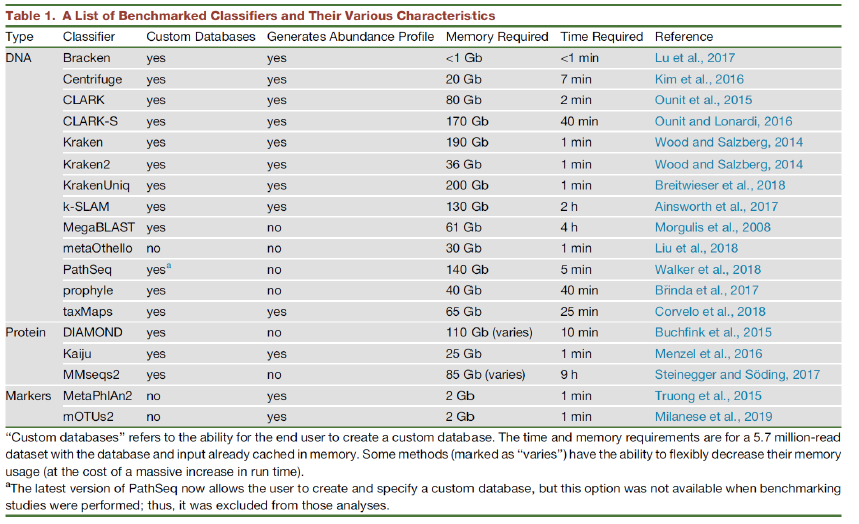
基本常用的软件都在上图中。一般DNA核酸比对还是使用Kraken2比较多,蛋白质一般使用DIAMOND和kauju,MetaPhlAn2也比较常用。
可以使用mock数据集进行测试。
三代测序数据处理
Centrifuge
这里主要介绍一下centrifuge软件,物种鉴定使用方式如下
1 | centrifuge -x centrifuge_h+p+v_20200318/hpv -U nanopore.fastq.gz --report-file report.tsv -S result.tsv -p 64 >centrifuge.log |
会输出两个结果
按照reads进行结果统计的结果centrifuge_output.tsv
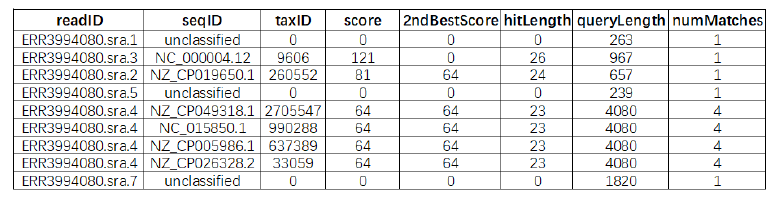
一共是八列,从左到右分别是reads ID、比对上的序列的Accession ID、物种分类ID、序列比对评分、第二比对结果的评分、比对上的部分的序列长度、总reads长度、该reads比对上的物种序列个数。
- 按照比对上的物种进行的统计centrifuge_report.tsv
.png)
一共是七列,从左到右分别是比对上的物种名字、物种分类ID、物种层级、基因组大小、比对上的reads数量(每一个reads可以比对上多个物种)、唯一比对上该物种的reads数量、丰度(比对上的区域/整个基因组的长度)。
在得到了结果之后就可以使用R等方式对tsv表格进行过滤。比如一条reads会比对到数据库中的多个物种,通常我们需要选择每条reads最优的比对,并且还要去除掉比对上的reads太少的物种。当然,命令行也能进行结果筛选。
1 | awk -F "\t" '{if ($3=="species" && $6 >5) print $1"\t"$6}' 0.01_report.tsv >0.01.txt |
这里标记一篇文章,Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection,这篇文章中有40个潜在肺炎病人肺部宏基因组测序的结果,可以通过Centrifuge比对的方式得到样品中占决定性多数的细菌种类,如果该细菌与肺炎有关那么就判定该患者有较大可能患有肺炎。该文章证明利用纳米孔基因组测序和宏基因组分析可以快速地进行临床诊断下呼吸道感染。其中的数据可以用来练习宏基因组物种分类。
二代测序数据处理
这一部分的几个软件或流程基本都来自于Biobakery workflow。这个流程来自于哈佛的Huttenhower团队。
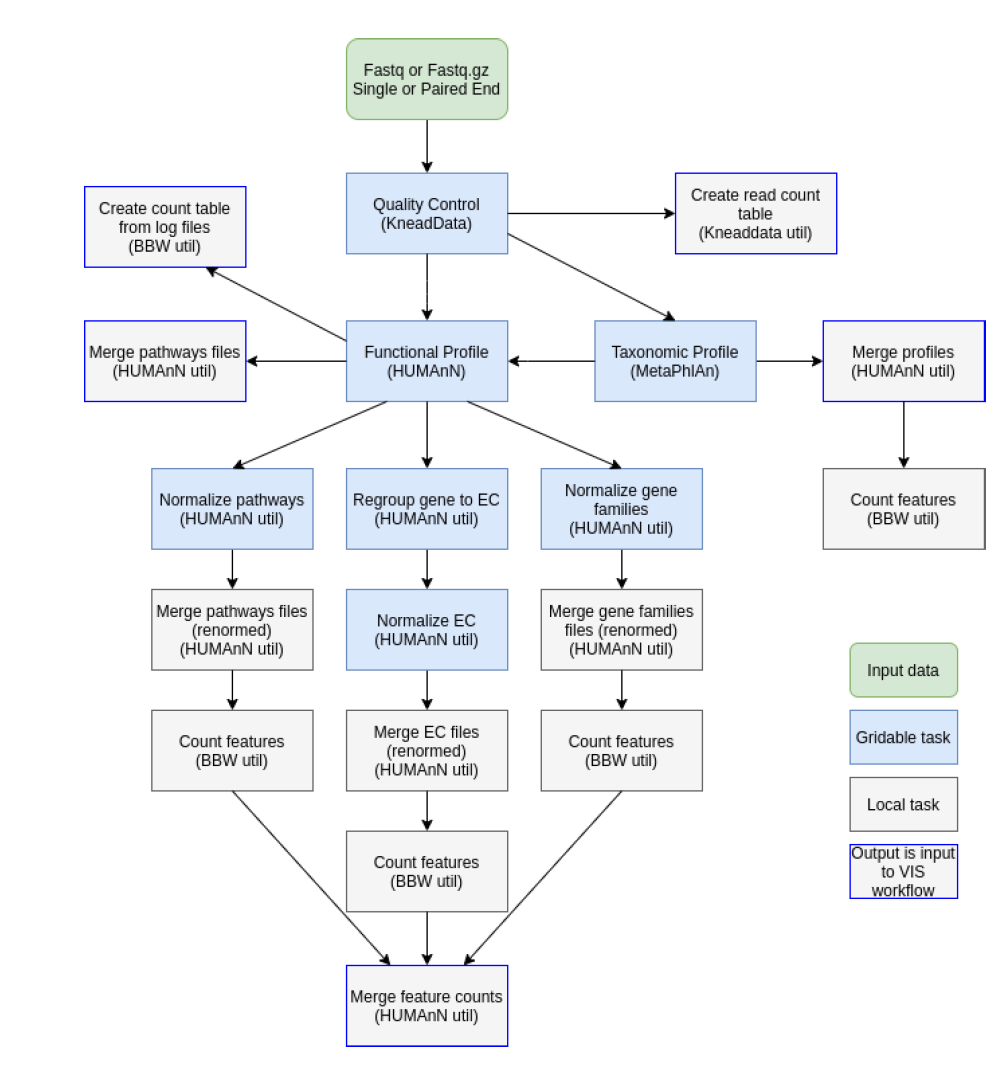
- 数据质控:使用Kneaddata,该软件先调用Trimmomatic过滤数据,然后利用bowtie2或bmtagger比对去除宿主数据。
- 如果不拼接数据,则可以直接使用MetaPhlAn2或者Kraken2实现序列的物种分类及物种丰度分析。功能鉴定则可以使用Humann完成。
- 也可以先对Reads进行拼接(Megahit、Metaspades等软件),然后使用Prokka软件从组装好的Contig或者Scafford预测基因,并使用cd-hit构建非冗余基因集。然后利用Nr、COG、GO、KEGG等数据库对基因进行注释。
- 如果有多样品则可以进行分组统计,将得到的物种/基因/功能表格利用R或者STAMP等软件在这三个层次进行分析。
Kneaddata
Kneaddata是一个数据质控过滤流程,整合了Fasqc质控,Trimmomatic数据过滤,Bowtie2比对数据库过滤宿主等功能。输入原始数据后就可以得到Cleandata。默认集成了人、小鼠、rRNA等数据库。如果是其他宿主,可以自行建库。
1 | kneaddata -i reads_1.fastq.gz -i reads_2.fastq.gz -db path\Homo_sapiens -o kneaddata_output --remove-intermediate-output -v -t 12 --trimmomatic-options 'ILLUMINACLIP:~/miniconda3/envs/biobakery/share/trimmomatic/adapters/TruSeq3-PE.fa:2:40:15 SLIDINGWINDOW:4:20 MINLEN:50' --reorder --bowtie2-options '--very-sensitive --dovetail' --run-fastqc-start --run-fastqc-end |
DB:/data/Food/primary/R0987_nextgen/Erkang.Zhang/20221130_course/course_4_meta/kneadData_database/
MetaPhlAn
MetaPhlan是用于二代测序物种分类的工具,利用快速比对工具Bowtie2与Marker基因集比对(非全基因组),所以运行速度非常的快。输入Kneaddata过滤后的数据就可以快速得到微生物群体中的物种组成。
MetaPhlAn调用Bowtie2比对,可以输入多种格式fasta、fastq、bowtie2out、sam等。
1 | metaphlan --input_type fastq --nproc 12 --bowtie2out metagenome.bowtie2.bz2 reads_filter.1.fastq.gz,reads_filter.2.fastq.gz -o SRS011243_abundance_table.txt |
结果导出的表格分为两列,第一列为物种分类,按照层级从大到小排列。第二列是物种的相对丰度,每一层级加起来都是100%,有些软件输出的则是reads数目。
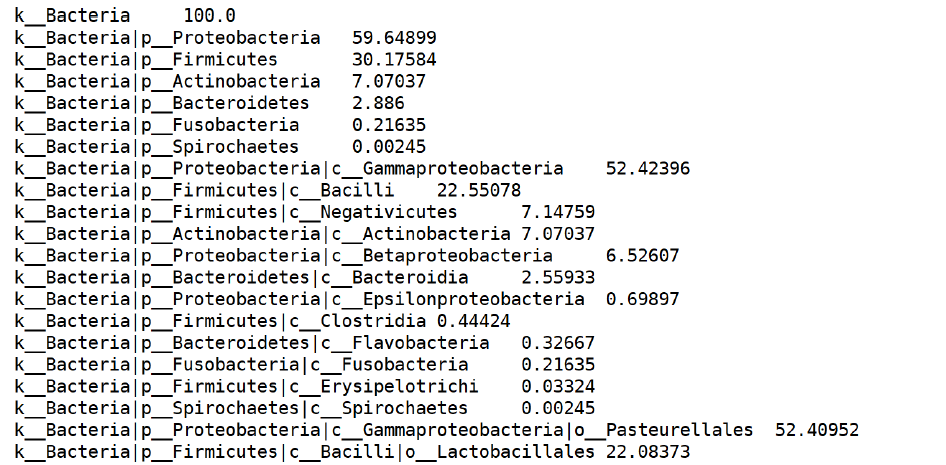
DB:/data/Food/primary/R0987_nextgen/Erkang.Zhang/20221130_course/course_4_meta/metaphlan_database/
HUMAnN
HUMAnN不仅可以通过调用MetaPhlAn完成物种组成部分还可以准确地获得微生物代谢途径和功能模块信息。
1 | humann --input-format fastq --input reads_filter.1.fastq.gz --input reads_filter.2.fastq.gz --output result --threads 12 --search-mode uniref90 |
结果会输出三个文件。
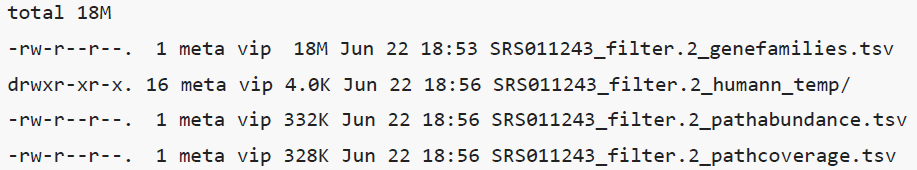
- 基因家族文件(genefamilies):群体中每个基因家族的丰度。基因家族是一组进化上相关的编码蛋白质序列,通常具有相似的功能。基因家族的丰度采用RPK表示(每kb的reads中该基因家族的含量),以此来标准化不同长度的基因家族的丰度。
- 通路丰度文件(pathabundance):代表群体中通路的丰度。
- 通路覆盖文件(pathcoverage)
另外,HUMAnM还支持比对到其他蛋白数据库的注释结果,以及对RPK的标准化/归一化处理,见该网站。
DB:/data/Food/primary/R0987_nextgen/Erkang.Zhang/20221130_course/course_4_meta/humann_database/
最终结果的可视化
Pavian可视化
Pavian是一款R包,但也有网页形式。可以用来可视化Kraken、MetaPhlAn、Centrifuge等软件的结果。如果要可视化Centrifuge的结果,需要先将其转化为Kraken的结果格式。
1 | centrifuge-kreport -x centrifuge_p/p_compressed result_log.tsv >kraken_result_log.tsv |
然后将其上传到网页上即可,可以绘制桑基图等。
Megan 可视化
Megan是一款综合性的微生物物种分类工具,除了支持物种分类还支持功能分析(KEGG数据库使用的是免费版本,付费版Megan可以使用全数据库)。支持多种格式输入,diamond比对的daa格式、blast比对格式、sam、biom等等。也可以用最简单的tsv、csv,两列即可,第一列物种名称、第二列物种丰度信息或reads数目。
GraPhlAn
也在Biobakery中,输入MetaPhlAn的结果可以直接绘制。
注意事项
- 现在的宏基因组分析流程基本都是先拼接生成Contig再继续分析物种、基因。